

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

2021年5月29日,美国食品和药物管理局(FDA)批准靶向抗癌药Lumakras(sotorasib,AMG510)上市,Sotorasib是一款高选择性的、不可逆转的KRASG12C突变抑制剂,是全世界第一款治疗KRAS基因突变癌症的靶向药。用于治疗携带特定的KRAS基因突变(KRAS-G12C),且用过至少一次其他药物(比如化疗、免疫疗法)的晚期非小细胞肺癌患者。
FDA基于总缓解率(ORR)和缓解持续时间(DoR)数据获得加速批准,后期的III期临床试验正在进行。
Kirsten大鼠肉瘤病毒癌基因同源物(KRAS)突变主要发生在吸烟人群中,在西方国家占肺癌发病率约20-25%,在亚洲约占10-15%。KRAS的变异主要发生在代码12(>80%)和13上,大约39%的变异为KRAS-G12C(甘氨酸GlycineG突变为半胱氨酸CysteineC),其他变异包括KRASG12V和KRAS-G12D等,吸烟人群和非吸烟人群有着不同的变异。KRAS变异的肺癌人群较野生型有较差的生存期(OS)。
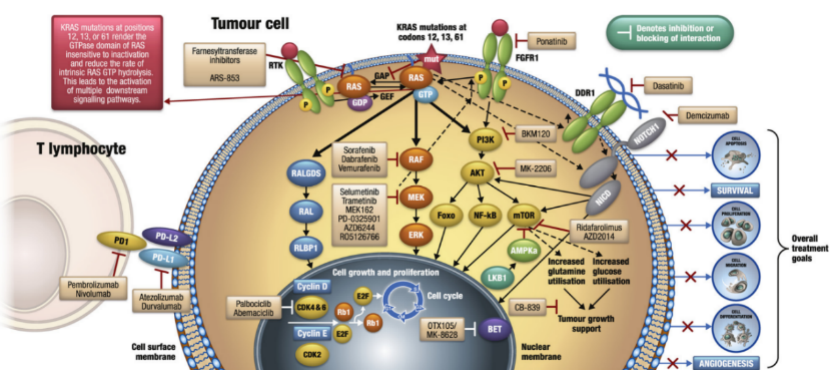

RAS蛋白通过不同的效应物调节,控制多种细胞功能的信号转导(RAS在这些靶点的上游),例如生存、增殖和分化。本构激活RAS癌蛋白在不存在的情况下启动细胞内级联反应细胞外信号,导致不受控制的细胞增殖和细胞存活异常。这些细胞功能的失调构成了癌症的许多标志。RAF是丝裂原活化蛋白激酶中的第一个激酶(MAPK)途径,使MEK磷酸化,进而激活细胞外信号调节激酶(ERK)。ERK都激活胞质底物并转移到细胞核以刺激参与细胞增殖、存活、分化和细胞周期调节。已经广泛证明MAPK信号通路在RAS介导的肿瘤发生。
磷脂酰肌醇-4,5-二磷酸3-激酶(PI3K)在RAS介导的肿瘤发生中发挥关键作用。激活时,PI3K通过磷酸化将磷脂酰肌醇4,5-二磷酸(PIP2)转化为磷脂酰肌醇3,4,5-三磷酸(PIP3),并且PIP3激活磷酸肌醇依赖性激酶1(PDK1),它在转磷酸化AKT,一种丝氨酸/苏氨酸特异性蛋白激酶。AKT激活导致其几种生理功能的磷酸化底物,如哺乳动物雷帕霉素靶蛋白(mTOR)、叉头boxO(FOXO)或核因子(NF)-κB,刺激细胞周期进展、存活、代谢、迁移和抵抗细胞凋亡。
RALGDS是另一种RAS效应器,其底物是RAS家族RAL-A/B小GTP酶。RALGDS也可以通过JunN-末端激酶(JNK)通路刺激转录促存活和细胞周期进程基因。这个信号通路在RAS介导的肿瘤发生中也有非常重要的作用。
KRAS是人类RAS基因家族的成员,编码一个小的GTPase膜结合蛋白,它可以存在于两种不同的状态:GDP结合,处于非活动状态,GTP结合,处于活动状态并通过与不同的下游效应器相互作用来转导信号。
突变的RAS癌蛋白在功能上发生改变,致癌形式阻止GAP增加GTPase的内在催化速率,从而将RAS保持在其组成性GTP结合的活性状态,从而激活致癌途径和细胞信号转导。
KRAS靶点被认为不可成药的靶点,有几个因素需要克服,野型KRas蛋白表面缺少小分子药物结合的较大的口袋。KRas和GTP高亲和力,无法竞争。另外,如果选择性较差,毒性会限制剂量。经过几年的努力,针对KRAS突变体的共价抑制剂研究的突破让通过异构位点(allosteric)靶向KRAS突变体成为可能。结构分析已揭示突变体的突变半胱氨酸12残基KRasG12C与存在的小口袋(P2)相邻仅在不活动的KRas-GDP状态下,这个表面凹槽成为最可能成药的靶点,广泛搜索与不可逆结合的共价抑制剂作为开发热点。
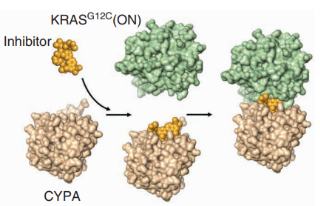
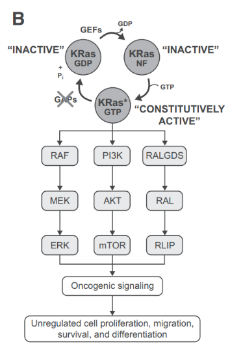
药物开发
Sotorasib是在ARS-160(华人科学家刘异博士研发)基础上对分子结构不断优化而来的,其与胱氨酸结合的烯烃特异性结构没有改变,增加了一些特异性位阻基团,选择性以及疗效较ARS-160更好。
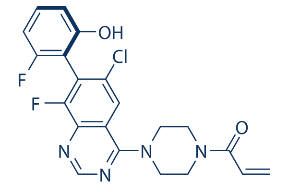
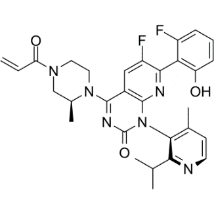
半胱氨酸的硫基侧链,它独特地使其能够与小分子抑制剂形成共价键。已经发现,ARS-1620的烯烃结构可以与硫键形成不可逆共价键。

不断的强化迭代筛选和基于结构的设计导致发现了小分子抑制剂,它们选择性地和不可逆地与在突变体KRasG12CP2口袋中半胱氨酸结合的并将其锁定在不活动的KRas-GDP状态。
其实,在治疗慢性淋巴细胞白血病的BTK抑制剂也是采用这个策略,这些结构下端都有相似的结构,可以与半胱氨酸的巯基发生共轭。但是一旦肿瘤蛋白发生突变,由半胱氨酸突变为其他丝氨酸(BTK会产生C481突变),活性以及选择性降低,会产生耐药性。

ARS-1620验证了KRAS(G12C)的直接抑制作用,但是进一步进行改良是困难的。因为ARS-1620占据的口袋体积小,为其他的蛋白质-配体相互作用提供了有限的途径,药效优化受到限制。
经结构分析,发现这个蛋白His95有一个另一种取向产生的表面凹槽,如被芳香环占据,会增强了与KRAS(G12C)蛋白的相互作用。AMG510作为His95沟结合分子优化的最佳候选药物。
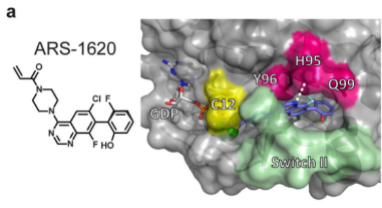
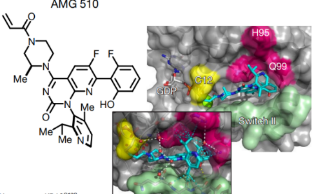
AMG510对KRAS(G12C)的占有率,在两种测定中均达到接近最大水平,约为0.2μM。AMG510还严重削弱了NCI-H358和MIAPaCa-2中的细胞活力(IC50分别为0.006μM和0.009μM,比ARS-1620强约40倍;
两种构型的IC50相差巨大,而且可以相互转换。

药物热稳定性差,构型会发生翻转,药效降低。构型对药效关系较大,在苯胺邻位加入基团,阻止翻转,对不同基团进行了筛选研究。
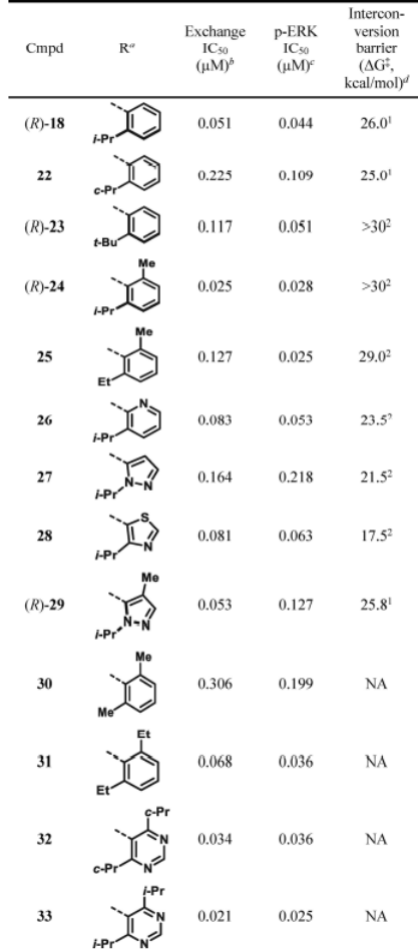
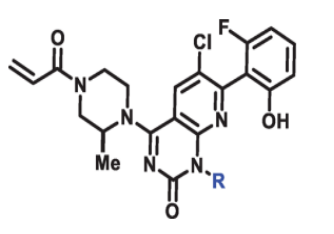
测定了不同基团的异构体相互转化的能量,邻位为异丙基和甲基以及异丁基基团时,翻转需要的能量最高。异丁基翻转的t1/2只有约5小时,不适合进一步开发。
在(R)-24表现出中等口服生物利用度(21%)和良好的体内靶标覆盖率(在Cmax时p-ERKIC50的覆盖率为4.5倍),25化合物28展示了更适度的体内靶标覆盖,并且化合物31-33未能显示出明显的体内靶标结合。

随后发现(R)-24的结晶物的相对于无定形形式口服生物利用度显着降低(4-12%)。两种策略进行结构改造:(1)改变喹唑啉酮C6卤素取代基(R部分),降低(R)-24的亲油性,和(2)在口袋结构环上(X和Y处)引入氮原子增加极性,而不改变药物活性。


双氮取代(36)嘧啶类似物增强了水中溶解度,但有较低的渗透性和口服生物利用度。接下来研究喹唑啉酮C6取代基是否降低其亲脂性、增强水溶解度同时保持细胞活性。C6氟代替氯取代基(化合物37)导致细胞活性降低,同时降低水溶性。但是,同时引入氮原子,可以导致显着增强的水溶解度。通过这些研究中,(R)-38作为候选药物。进一步研究(R)-38的PK/PD关系,结果表明,ERK磷酸化的最大抑制是达到给药后60-120分钟,(R)-38血浆峰值是给药后30分钟,在120分钟时间点,(R)-38的浓度较低。

在具有人类肿瘤细胞MIAPaCa-2T2(p.G12C)小鼠中,肿瘤每天增长约86%,每天给药10-100mg/kg,10mg/kg的(R)-38显著抑制了肿瘤,在≥30mg/kg的剂量下肿瘤消退。

实现MIAPaCa-2T2肿瘤消退所需的(R)-38剂量比ARS-1620低至少3.3倍。基于令人信服的及其显着的治愈KRASp.G12C突变体的能力以及抗体内肿瘤药理学特征,(R)-38是具有前途的药物,开始对(R)-38作为AMG510用于临床开发。
使用Sprague-Dawley大鼠和Beagle犬评估了安全性长达3个月的重复剂量毒理学研究。在这两个物种中sotorasib治疗导致胆固醇增加和红细胞(RBC)减少与骨髓组织学发现无关的参数。肝脏是大鼠高剂量胆红素、AST和ALT升高的常见目标和增加的胆红素和碱性磷酸酶、肝脏重量增加和小叶中心肝细胞肥大暴露量约为960mg人体剂量下临床AUC的0.4倍。较高的ALT/AST也在临床上观察到,LUMAKRAS标签包括肝脏升高的警告。
AMG510于2018年8月进入人体临床试验,评估其在KRASp.G12C突变肿瘤中的安全性、耐受性、药代动力学特性和疗效的I/II期试验(NCT0360088334)。早期结果研究证实AMG510表现出色晚期KRAS患者作为单一疗法给药时的耐受性和有希望的抗肿瘤活性p.G12C突变实体瘤。
采用双参数贝叶斯逻辑回归模型(BLRM/BOIN设计)剂量爬坡试验设计,不限定入组人数,比传统的3+3方法效率更高。剂量组分别为180,360,720,和960mg四个剂量。

由于没有确定适应症,试验纳入了包括59例非小细胞肺癌,结直肠癌42人,其他肿瘤类型28人(篮式设计),可以看出开发者主要是想在NSCLS(KRASp.G12C突变高表达)方面有所突破。
DLT确定:根据CTCAEV5.0制定
血液学毒性-发热性中性粒细胞减少症-中性粒细胞减少感染-4级中性粒细胞减少症-≥3级血小板减少症持续>7天-3级血小板减少症伴≥2级出血-4级血小板减少症-4级贫血·非血液学毒性-≥4级呕吐或腹泻-3级腹泻或3级呕吐持续超过3天,尽管最佳医疗支持-尽管有最佳医疗支持,但≥3级恶心持续3天或更长时间-任何其他≥3级不良事件。
试验结果:960mg剂量没有达到MTD。
有效性:客观反应:肺癌32.2%,直肠癌7.1%,其他癌14.3%。
疾病控制:肺癌88.1%,直肠癌73.8%,其他癌75.0%,在治疗非小细胞肺癌方面表现较其他癌种要好很多,故下一步重点在非小细胞癌种进行临床研究。
非小细胞癌方面的研究-NCT03600883,采用I/II期无缝设计,采用BLRM试验设计,提高了RP2D的准确性,同时也加快了剂量爬坡的速度。研究包括多剂量耐受性,PK/PD研究,食物影响,确定了有效剂量以及有效性。
1)药物代谢 
以上药代数据显示每个剂量组的药代数据接近,没有区别。高剂量组没有出现较高的暴露量。FDA认为960mg剂量组并非最优剂量,Sotorasib的BCSII类,其药剂处方仍有优化的余地。
2)有效性临床试验
试验设计
受试者:病理记录显示局部晚期或转移性恶性肿瘤;通过DNA测序鉴定出KRASp.G12C突变。受试者必须接受过适合其肿瘤类型和疾病阶段的先前标准治疗,或者在研究者看来,不可能耐受或从适当的护理标准治疗中获得临床上有意义的益处。给药剂量为每天960mg;
主要终点是由盲法、独立、影像学审查的客观反应(完全或部分反应)
次要终点指标为应答持续时间,疾病控制,应答时间,疾病进展以及生存率以及安全性。
样本量考虑(单臂试验设计):
在所有2线治疗NSCLC的ORR中雷莫芦单抗联合多西他赛疗效最好。

雷莫芦单抗联合多西他赛作为晚期NSCLC患者二线治疗试验结果显示,最终ORR为23%。基于SAD试验的32%ORR,把握度90%,95%置信区间下限高于23%,需要受试者105例。
在126名入组患者中,大多数(81.0%)接受过铂类化疗和PD-1或PD-L1治疗。124名患者有基线时可测量的疾病,并评估反应。在46名患者中观察到客观反应(37.1%),其中4人(3.2%)有完全反应,42人(33.9%)有部分反应。中位缓解持续时间为11.1个月。100名患者(80.6%),中位无进展生存期为6.8个月,中位总生存期为12.5个月。126例中有88例发生了与治疗相关的不良事件患者(69.8%),包括25名患者(19.8%)的3级事件和4级事件事件1(0.8%)。客观反应为37.1%,达到了预期。
Sotorasib常见的所有等级的副作用和不良反应有:腹泻(42%)、肌肉骨骼疼痛(35%)、恶心(26%)、疲劳(26%)、肝毒性(25%)、咳嗽(20%)、呕吐(17%)、便秘(16%)、呼吸困难(16%)、腹痛(15%)、浮肿(15%)、食欲变差(13%)、关节痛(12%)、肺部感染(12%)、皮疹(12%)。
腹泻发生率还是较高的,这是大部分口服小分子靶向药物(不同靶向药物如,EGFR、VEGFR以及PI3Kδ等)以及卡瑞利珠单抗、尼妥珠单抗、帕博利珠单抗和信迪利单抗(免疫药物注射)等单克隆药物共同的副作用。目前,临床上尚无治疗分子靶向药物相关性腹泻的特效药物,仍在沿用以往用于治疗化疗性相关腹泻的洛哌丁胺等药物。分子靶向药物相关性腹泻的发病机制目前尚不明确,可以看出与靶点以及是否是口服无关,然而这种不良反应在动物安全性试验的报道较少,是人类特异性原因还是神经方面等其他因素,目前不得而知。
上市前,申请人与Guardant和Qiagen合作提交Guardant360CDx试剂盒(P200010/S012)和QIAGENtherascreen®KRASRGQPCR试剂盒(P110027/S012)批准申请(PMA),分别用于定性检测KRASG12C基因突变(用血细胞检测即可,无需活检样本)。
化合物专利:作为用于治疗肺癌、胰腺癌或结直肠癌的KRASG12C抑制剂的苯并异噻唑、异噻唑并[3,4-b]吡啶、喹唑啉、酞嗪、吡啶并[2,3-d]哒嗪和吡啶并[2,3-d]嘧啶衍生物;
中国申请号201780087190.7,目前处于公开状态,公开号CN110366550A。
授权分析:占据His95表面凹槽的特异性分子结构,以及改变生物利用度以及溶解度分子结构部分可以授权,其他结构不具有新颖性。
小结
Sotorasib在ARS-1620基础上对化学结构上进行了改造,其选择性以及疗效更佳。采用高效快速的临床试验设计,Ia期采用BOIN以及篮式试验设计,快速确定适应症以及最佳有效剂量。Ib期MAD与单臂的有效性试验完美相结合,确定了PK、PD以及有效性指标ORR,最终临床试验结果显示,在ORR上完胜雷莫芦单抗联合多西他赛治疗方法。后期III期临床试验正在进行。
Sotorasib针对KRAS的靶向药的开发,具有里程碑式的意义,其在肿瘤界的影响力可以与伊马替尼媲美。RAS基因被发现是肿瘤中最常见的突变基因之一,相信在后期会有更多的此靶点的抗肿瘤药物发现和上市。
参考文献:
1、Irene, Ferrer, Jon, et al. KRAS-Mutant non-small cell lung cancer: From biology to therapy.[J]. Lung Cancer, 2018.
2、Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019 Nov;575(7781):217-223. doi: 10.1038/s41586-019-1694-1. Epub 2019 Oct 30. PMID: 31666701.
3、Lanman B A , Allen J R , Allen J G , et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors[J]. Journal of Medicinal Chemistry, 2019,
4、 Hong D S , Fakih M G , Strickler J H , et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors[J]. New England Journal of Medicine, 2020, 383(13).
5、Skoulidis F , Li B T , Dy G K , et al. Sotorasib for lung cancers with KRAS P.G12C mutation[J]. New England Journal of Medicine, 2021, 384(25).
免责声明






