

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!
撰文|《医药研发达人》 日语主编 高野哲臣
缘起
自我介绍

中国药品监管变革


八个方面的总结
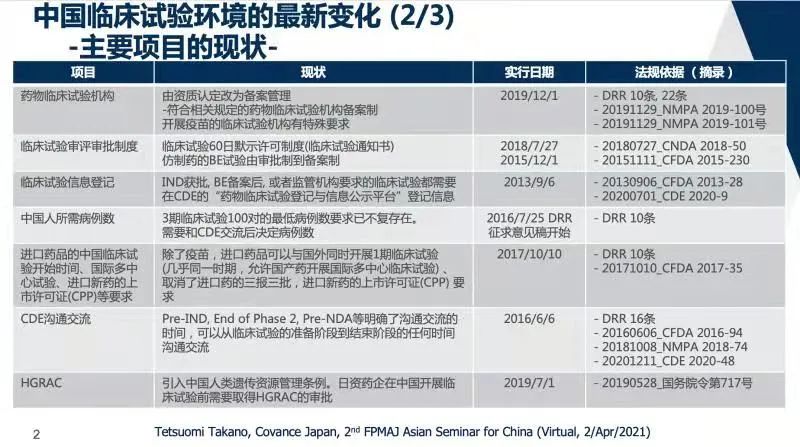
2015年11月11日,原CFDA发布《关于药品注册审评审批若干政策的公告(2015年第230号)》 ,该公告规定,自2015年12月1日起, BE试验由审批制改为备案制。
(6) 中国制药企业新药研发能力的演进与提升(从仿制药到小分子新药,再到抗体药物、基因治疗、细胞治疗/再生医学产品等新形态)
2011年11月1-2日,在东京一桥举行的APEC MRCT Tokyo Workshop(亚太经合组织国际多中心临床试验东京研讨会)上,中国CDE化药临床一部部长杨志敏介绍了表皮生长因子受体(EGFR)酪氨酸激酶抑制剂埃克替尼从I期临床试验的IND申请及开展,到2011年6月NDA获批历时5年半的临床开发历程。
结语
医薬研発達人微信公众号:

医薬研発達人日语网站:
http://www.pharmadj.com/member/jp/regist_step1.htm
日本读者请搜索ID:@707mfqir 关注医薬研発達人LINE公众号
我们的“编辑长”
上个月,研发客推出了国内和日本首家报导中日医药行业的媒体——《医药研发达人》主编高野哲臣先生的发刊词,本期我们特别将其编译成中文,以飨国内读者。
两个月以来,我与志同道合的朋友为这一新专栏不遗余力的撰稿、翻译、编辑、出版,在这一过程中,也有幸近距离接触工作中的高野先生。在此写下对他的个人印象一文,作为编后花絮。
自担任《医药研发达人》日语版“编辑长”(日语:主编之意)以来,高野哲臣先生就多了一个新名字,因为,日本编辑部的同事植村老师、琳琳等和我都亲切的喊他“编辑长”。
我们的编辑长具备很高的专业水平。他精通中日两国和亚太国家的药政法规和临床开发;他会第一时间告诉你,为何我国药监局能在4年后成功连任ICH管委会成员;他分析,中国的药物创新,之所以取得今天的成就,是源于过去数十年厚积薄发的药品监管改革,及举国上下的资本汇聚和制药行业的人才济济;他指出,中日制药行业药品研发合作前景广阔,但欲速不达,所有中日申报的临床研究数据需基于ICH框架下加以科学的审评和决策。
我们的编辑长工作认真负责。他每天工作至深夜2点,我们常在凌晨2-3点收到他的邮件。不过,他总能在百忙中为我们开选题会,指导报导方向,提供采访资源,把关药品监管和制药技术、中日之间的政治和文化问题。
我们的编辑长做事精益求精。他在稿件终审时,逐字逐句每一标点符号细细修改,并注明原因,同时将所有修订制定成一个表格供我核对。他至今仍收藏多年前参加过的会议日程、PPT。每一天在互联网上浏览的中国和全球医药信息,都被他分门别类的亲自整理,归档好在自己的电脑里。

图为高野哲臣制作的校对清样表。
我们的编辑长思维缜密严谨。他在发表意见之前总会紧锁眉心,俯首低头,经过一番深思熟虑以后,才会将他的观点抛出,但往往点到之处,都是重中之重的建议。当你问他每一个答案应“是”还是“否”,他会详细列出是和否的理由,而不会理所当然给你建议。
我们的编辑长对同事要求很严格。我不止一次被他责备,那是因为我粗枝大叶的工作而导致。这时候,他会像前辈一样教导我,希望我能在每一次采访前多做准备,希望我能写出一篇又一篇客观公正、内容详实的报导。他常常对我说,要建立在行业中的名声度、信誉度和品牌知名度,树立中国记者良好、权威和专业的形象。对他的谆谆教诲,我倍感珍惜,永志不忘。
我们的编辑长深爱着中国。他自2003年开始关心中国药监改革和制药行业,并组织了不下50多场有关中国的演讲,在每一次演讲前,他会想象每一个观众的面庞,准备好他们关心的话题,希望做到最专业的演讲。
当我问他,高野先生,您作为一位日本专家却如此关心中国,您是否在意身边的同事和日本同行如何看待你?这时,他语重心长的对我说,每当他内心涌现出一丝不确定或恐惧时,他会像村上春树一样去长跑,要么泡个澡,或者干脆不理会别人的评论而倒头大睡。他强调,他从不放弃自己心中的理想,也不曾间断对中国法规和行业知识的学习。他会一次又一次地完成自己设定的目标,并与中国专家保持交流。
“我要通过自己的努力赢得大家的认可和尊重。而每当发觉自己取得了一些进步,我的恐惧和担忧就会减少一些,进而更坚定自己的目标,勇往直前。”他说。
事实证明,他多年前对中国制药监管创新的预判都是正确的。中国今天已成为全球第二大医药市场、努力向国际发达监管国家看齐的制药大国。而他在中国也收获了丰盛的友谊,最近还成为我们《医药研发达人》的主编。正如他在发刊词里写道:“国家药监局(追求创新和监管改革的)想法坚定不移,是具有可持续性、可预见性和稳定的。《医药研发达人》将为中日医药行业之间架一座桥梁,是我内心质朴无华和强烈的愿望。”
这就是我们的编辑长高野哲臣先生,我们十分敬爱他,希望在他的指引下,《医药研发达人》这一富有使命感的事业大展宏图,也祝愿中日两国合作与友谊万古长青!
(冬蕾)







