

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

责编 | 兮
蛋白质是细胞功能的基本执行者,而不同蛋白之间的相互作用(protein-protein interaction,PPI)则是蛋白执行其相应功能的主要方式之一。明确某一基因所编码之蛋白与其他蛋白组成员之间的物理关联对于理解这一基因与其它基因间的功能联系具有极为重要的意义,前者在某种程度上甚至可视为后者的直接逻辑前置【1】。为了准确测定PPI的存在及亲和度,人们开发出了多种分子生物学实验手段,包括但不限于酵母双杂交(yeast two-hybrid,Y2H)、亲和纯化-质谱联用(affinity-purification mass spectrometry, AP/MS)、蛋白共纯化(co-purification)、BioID/APEX邻近标记技术,及荧光共振能量转移(FRET)等。
不过,由于蛋白质数目之庞杂、可变体结构特征之繁复、蛋白翻译后修饰种类之丰富,及PPI本身所具有的高度动态和环境依赖特征,上述实验方法所能应用之尺度十分有限。为此,人们同时开发了各类用于推测PPI的计算分析方法,包括基于基因共表达、功能共富集、文本挖掘、或机器学习/深度学习预测的各类手段。这类方法在可靠性上尽管无法媲美实验证据,却能够大规模、系统性地识别全蛋白组水平的潜在PPI,因而在蛋白组学研究中亦具有十分重要的地位。在实验证据和计算预测的双重支持下,人类对于PPI的认知已经达到了百万甚至千万数目的层级;以STRING和BioGRID等为代表的综合性、大规模数据库收录了基于各类实验或预测支持的PPI数据,并进行了严格的分类与质控,成为研究界广泛参考的重要资源【2】。
2021年7月1日,来自加拿大英属哥伦比亚大学(University of British Columbia)的Leonard J. Foster和Joerg Gsponer在Cell上发表了题为的An atlas of protein-protein interactions across mouse tissues的研究,报道了首个小鼠多器官全蛋白组尺度互作图谱,为生物蛋白互作组研究提供了最新的重要助力。

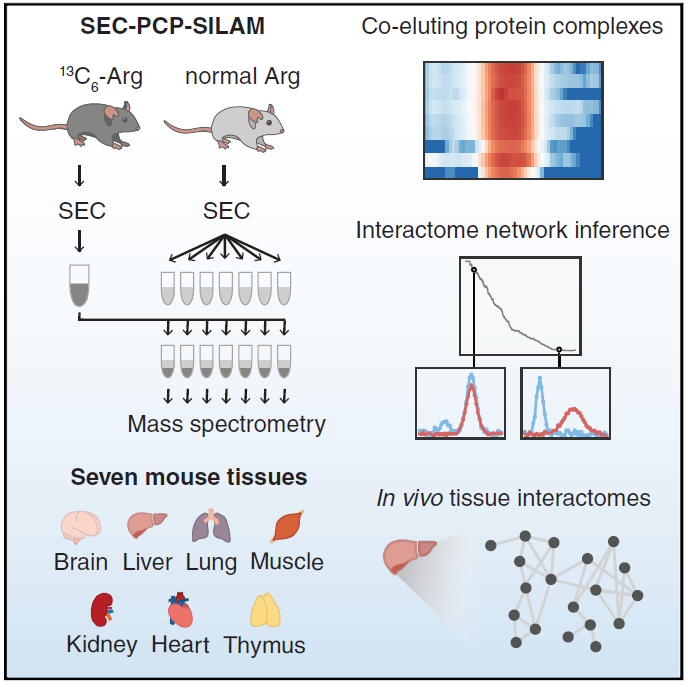
在该研究中,作者利用SEC-PCP-SILAM方法,即尺寸排除色谱法(size exclusion chromatography,SEC)、蛋白关联分析(protein correlation profiling, PCP)和哺乳动物总体蛋白质稳定同位素代谢标记(stable isotope labeling of mammals,SILAM)三者的结合,对小鼠的七种组织——脑、肝、肺、肌肉、肾、心脏和胸腺进行了分析。这一方法之所以适用于研究蛋白质互作,原理在于其能够对单一组织样本中的蛋白质复合物依据理化性质的差异进行分群捕获和定量,从而通过不同蛋白分子在从单一组织样本中得来的数十个群落之间的协同变化程度来反映互作关系的强弱。具体到本研究中,研究者将每个小鼠组织分为了55个群落,并进行了重复组分析。
如前所述,在高通量蛋白组学技术的帮助下,研究界对哺乳动物蛋白互作组的描绘已经达到了接近全蛋白组的尺度。然而,在横向维度上,即组织或细胞类型的丰富性上,这一努力则显得欠缺了许多。例如,迄今最大规模的人体蛋白互作图谱BioPlex,即是基于永生化细胞系而非真实组织或原代细胞产生的。因此,这些在单一非生理环境中捕获的蛋白互作关系有多少能够迁移到真实的病理生理研究场域中去为相关分子机制提供支持,就成了一个亟待回答的疑问。
显然,该研究凭借对多样化的真实组织的蛋白互作关系进行捕获,为解决这一困难提供了一个极佳的入口。例如,作者通过对比分析发现,以往常被研究界采用的,通过基因表达的组织特异性特征来预测相应蛋白是否在某一缺乏直接蛋白组证据的组织或细胞类型中具有某一特定互作关系的算法,在很大程度上是不可靠的。具体而言,在很多情况下,虽然编码一对蛋白分子的两个基因的表达水平在不同组织中表达程度并无较大差异,但这对蛋白分子却仅在部分组织中呈现互作关系。这无疑提示了基因差异表达这一要素对于蛋白互作存在性这一结果的影响权重可能是十分有限的,因此不宜被过度依赖。
基于该研究所提供的宝贵资源所能回答的另一个有趣的问题是,组织特异性越弱的蛋白互作关系是否在分子进化上也倾向于更加保守。为了研究这一问题,作者收集了跨越较长进化尺度的多个物种中现有的蛋白互作数据,然后以本研究中所捕获的蛋白互作网络进行了对比分析,结果发现在7种组织中都存在的蛋白互作关系在三种低等生物中(线虫、果蝇、酵母)具有最高的复现率,在6种中存在的稍低,以此类推。同时,编码那些参与弱组织特异性互作关系(即在多数组织环境中都存在的互作关系)的蛋白分子的基因在进化树上也处于更为早期的位置。
最后,作者试图从基因功能和蛋白特性角度探究对某一蛋白分子的“朋友圈”(即与之存在互作关系的所有其它蛋白分子)的组织特异性强弱的影响因素。结果发现,那些在不同组织环境中都具有稳定互作伙伴的蛋白分子显著地拥有更少的内在无序(intrinsic disorder)结构、更少的线性基序(linear motif)和更少的磷酸化修饰。另外,作者还发现已知与疾病相关联的基因所编码的蛋白显著地富集于“朋友圈”组织特异性强的类群。
总之,这一研究利用高通量定量蛋白组学技术首次绘制了哺乳动物真实生理环境中的蛋白互作图谱,并凭借对多样性生物背景的覆盖实现了对保守与非保守蛋白互作关系的区分,以及对这种区分所蕴含的分子机制与生物学意义提供了积极的探索和良好的解释,因此其超越了作为一项生物学资源的单纯意义,而成为了不可多得的生物学机制研究的典范。
原文链接:
https://doi.org/10.1016/j.cell.2021.06.003
参考文献
1. de Las Rivas, J. & Fontanillo, C. Protein-protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 6, 1–8 (2010).
2. Bajpai, A. K. et al. How helpful are the protein-protein interaction databases and which ones ? bioRxiv 1–33 (2019) doi:10.1101/566372.
3. Rolland, T. et al. A proteome-scale map of the human interactome network. Cell 159, 1212–1226 (2014).
4. Huttlin, E. L. et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162, 425–440 (2015).
5. Huttlin, E. L. et al. Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509 (2017).
6. Go, C. D. et al. A proximity biotinylation map of a human cell. bioRxiv 3, 58–66 (2019).
7. Luck, K. et al. A reference map of the human binary protein interactome. Nature 580, 402–408 (2020).
8. Huttlin, E. L. et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022-3040.e28 (2021).
转载须知
【原创文章】BioArt原创文章,欢迎个人转发分享,未经允许禁止转载,所刊登的所有作品的著作权均为BioArt所拥有。BioArt保留所有法定权利,违者必究。







