▎药明康德内容团队编辑
最常见非霍奇金淋巴瘤闻“噩耗”!FDA加速批准“加强版”抗体疗法上市
MorphoSys和Incyte联合宣布,美国FDA已加速批准Monjuvi(tafasitamab-cxix)上市,与来那度胺(lenalidomide)联用,二线治疗成人复发/难治性弥漫性大B细胞淋巴瘤(DLBCL)患者,包括低级别淋巴瘤引起的DLBCL,以及不适合接受自体干细胞移植(ASCT)的患者。Monjuvi是一种通过改造抗体Fc端增强细胞介导的细胞毒性反应的人源化抗CD19单克隆抗体,这一批准是根据总缓解率(ORR)获得的加速批准。
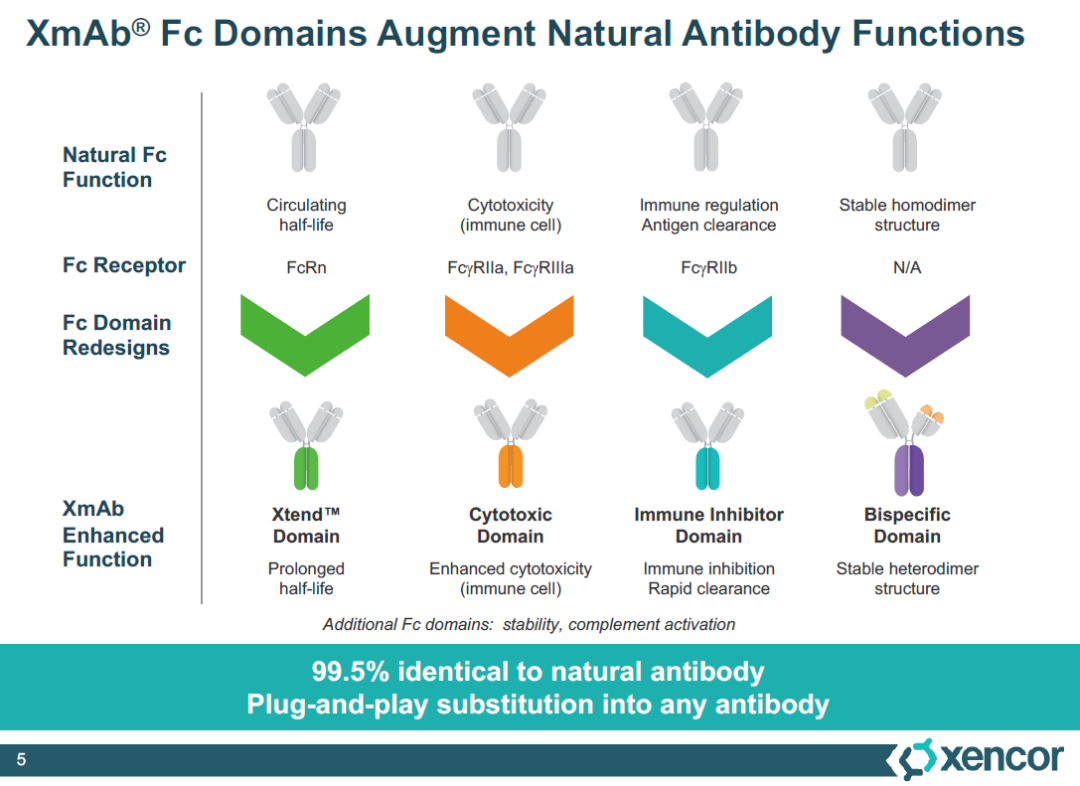
2010年,MorphoSys从Xencor获得了开发tafasitamab的全球独家权益。Tafasitamab利用了名为XmAb的Fc结构域改造技术,通过改造抗体的Fc端,能够将抗体与Fcγ受体的亲和力提高40倍,从而更好地激活先天杀伤细胞(NK cells)、巨噬细胞等免疫细胞,通过抗体依赖性细胞介导的细胞毒性(ADCC),和抗体依赖性细胞吞噬作用(ADCP)介导B细胞裂解。FDA此前授予Monjuvi和来那度胺联合治疗复发/难治性DLBCL的快速通道资格、突破性疗法认定和优先审评资格。
▲XmAb技术平台能够通过改造Fc结构域增强抗体的多种特征(图片来源:Xencor官网)
FDA的批准是基于MorphoSys进行的开放标签、多中心、单组2期临床试验L-MIND的数据。研究结果显示,Monjuvi与来那度胺联用,达到55%的总缓解率(ORR),包括37%的完全缓解率和18%的部分缓解率。中位缓解持续时间(mDOR)为21.7个月。
24小时内改善抑郁症状,杨森创新疗法获批治疗有自杀倾向抑郁症患者
强生(Johnson & Johnson)旗下杨森(Janssen)公司宣布,美国FDA已批准Spravato(esketamine)CIII鼻喷雾剂的补充新药申请(sNDA),与口服抗抑郁药联用,治疗伴有急性自杀念头或行为的抑郁症(MDD)成人患者的抑郁症状。新闻稿指出,Spravato是第一个也是唯一一个FDA批准的在24小时内显示减轻抑郁症状的药物,为显著缓解症状提供了一个新选择。
Spravato鼻喷剂是一种非选择性、非竞争性NMDA受体拮抗剂。它具有一个与目前可用的抑郁症药物不同的新作用机制。
去年,它获得FDA批准治疗成年治疗抵抗性抑郁症(treatment resistant depression)患者。
这一适应症的获批,是基于两项完全相同的3期临床试验,数据显示,Spravato加综合标准治疗在24小时内能够显著、快速减轻抑郁症状。一些患者最早在服药4小时就开始出现应答。在两项试验中,给药后24小时,Spravato加综合标准治疗导致MADRS抑郁评定量表(一种用于评估抑郁症状严重程度的工具)评分下降了15.9和16.0分。相比之下,安慰剂加综合标准治疗组降低了12.0和12.2分。
Spravato组和安慰剂组患者在服药后4小时到25天期间症状持续改善,在两项试验中,Spravato组有41%和43%达到抑郁症的临床缓解(轻微或无症状),相比之下,在双盲期结束时,安慰剂组有34%和27%达到抑郁症的临床缓解。
有望迎来第三项适应症!诺华CAR-T细胞疗法临床结果亮眼
诺华(Novartis)公司宣布,其CAR-T细胞疗法Kymriah(tisagenlecleucel),在治疗复发/难治性(R/R)滤泡性淋巴瘤(FL)患者的2期临床试验ELARA中获得积极结果。中期分析显示,这一全球性研究达到了独立审查委员会评估的完全缓解率(CRR)的主要终点。ELARA试验的结果将在即将召开的医学会议上公布,并将纳入美国和欧盟监管申请资料中。
滤泡性淋巴瘤是非霍奇金淋巴瘤(NHL)的第二常见类型,是一种惰性淋巴瘤,约占NHL病例的22%。尽管有治疗方法可改善总生存期,但FL被认为是不可治愈的恶性肿瘤,呈现不断复发和缓解的疾病进展模式。在复发FL患者的一生中,他们可能需要接受5种以上的不同治疗,最高可达到12种。此外,由于这种复发和缓解的疾病进展模式,对治疗没有应答或快速复发的患者可能会穷尽可用的治疗方案。
Kymriah是FDA批准的首个靶向CD19抗原的CAR-T细胞疗法,也是首次在两个截然不同的适应症中获得批准的CAR-T疗法。这是一种一次性治疗,旨在增强患者的免疫系统对抗癌症的能力。Kymriah目前已获批用于治疗R/R急性淋巴细胞白血病(ALL)患者,以及R/R弥漫性大B细胞淋巴瘤(DLBCL)成人患者。
默沙东(MSD)和韩美(Hanmi Pharmaceutical)共同宣布,两家公司已经就用于治疗非酒精性脂肪性肝炎(NASH)的在研疗法efinopegdutide(以前称为HM12525A)的开发、生产和商业化达成了一项独家许可协议。Efinopegdutide是韩美开发的每周一次的胰高血糖素样肽-1/胰高血糖素受体(GLP-1/GCGR)双重激动剂,拟用于治疗非酒精性脂肪性肝炎(NASH)。先前已在多项1期和2期临床试验中评估了efinopegdutide的安全性和有效性,包括治疗伴有和不伴有2型糖尿病的重度肥胖患者。
根据协议,默沙东将获得efinopegdutide在美国和全球开发和推广的独家许可;韩美将获得1000万美元的前期付款,并有资格获得高达8.6亿美元里程碑付款,以及获批产品销售额的分成。韩美保留efinopegdutide在韩国商业化的选择权。
一线治疗非小细胞肺癌!辉瑞第三代ALK抑制剂达到3期临床终点
辉瑞(Pfizer)公司宣布,该公司开发的第三代ALK抑制剂Lorbrena(lorlatinib),在治疗初治晚期ALK阳性非小细胞肺癌(NSCLC)患者中开展的3期临床试验CROWN中达到了主要终点。该研究显示,与ALK抑制剂活性对照相比,Lorbrena(lorlatinib)显著改善了患者的无进展生存期(PFS)。CROWN的研究结果将提交给即将召开的医学大会。
肺癌是全世界癌症相关死亡的首要原因。非小细胞肺癌约占肺癌的80%-85%,约3%-5%的非小细胞肺癌病例携带ALK阳性肿瘤。在获得靶向治疗和免疫治疗之前,晚期NSCLC患者的五年生存率仅为5%。
Lorbrena在携带ALK重排的临床前肺癌模型中显示出高度活性。Lorbrena专门开发用于抑制对其他ALK抑制剂耐药的ALK基因突变,并可穿透血脑屏障。
2018年,美国FDA加速批准Lorbrena治疗经治ALK阳性转移性NSCLC患者。CROWN临床试验为这一加速批准的验证性试验。基于这一试验的积极结果,辉瑞将向FDA寻求将加速批准转变为完全批准,并且寻求批准治疗初治ALK阳性转移性NSCLC患者。
迎击多发性骨髓瘤!FDA加速批准首个BCMA靶向疗法
葛兰素史克(GSK)宣布,美国FDA已批准抗体偶联药物(ADC)Blenrep(belantamab mafodotin-blmf)作为单药疗法,用于既往接受过至少4种疗法(包括抗CD38单克隆抗体、蛋白酶体抑制剂和免疫调节剂)的复发/难治性多发性骨髓瘤成人患者。根据总缓解率,该适应症获得加速批准。Blenrep是全球首个获得批准的靶向B细胞成熟抗原(BCMA)的疗法。
Blenrep是一种抗体偶联药物,由人源化抗B细胞成熟抗原(BCMA)单克隆抗体和细胞毒药物澳瑞他汀F(auristatin F)通过不可切割的连接子偶联而成。它曾获得了FDA的突破性疗法认定和优先审评资格。值得一提的是,该产品已于今年5月在中国获得临床试验默示许可,适应症为:联合硼替佐米和地塞米松,用于治疗至少接受过一种既往治疗的多发性骨髓瘤成人患者。
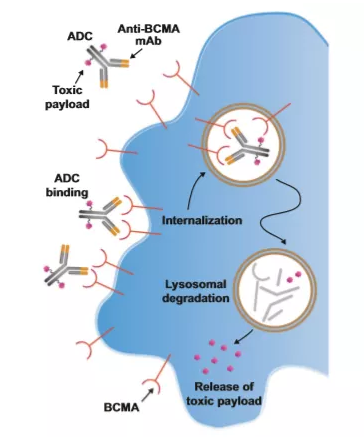
▲靶向BCMA的ADC作用机制(图片来源:Leukemia)
Blenrep的获批,是基于DREAMM-2研究的6个月初步结果,该研究纳入了复发/难治性多发性骨髓瘤患者,这些患者尽管接受了标准治疗,但病情仍在恶化。试验结果表明,在中位接受过7种前期治疗的患者(n=97)中,Blenrep的总缓解率(ORR)为31%(97.5% CI;21-43);随访6个月时尚未达到中位缓解持续时间(DoR),73%的缓解患者的DoR≥6个月。
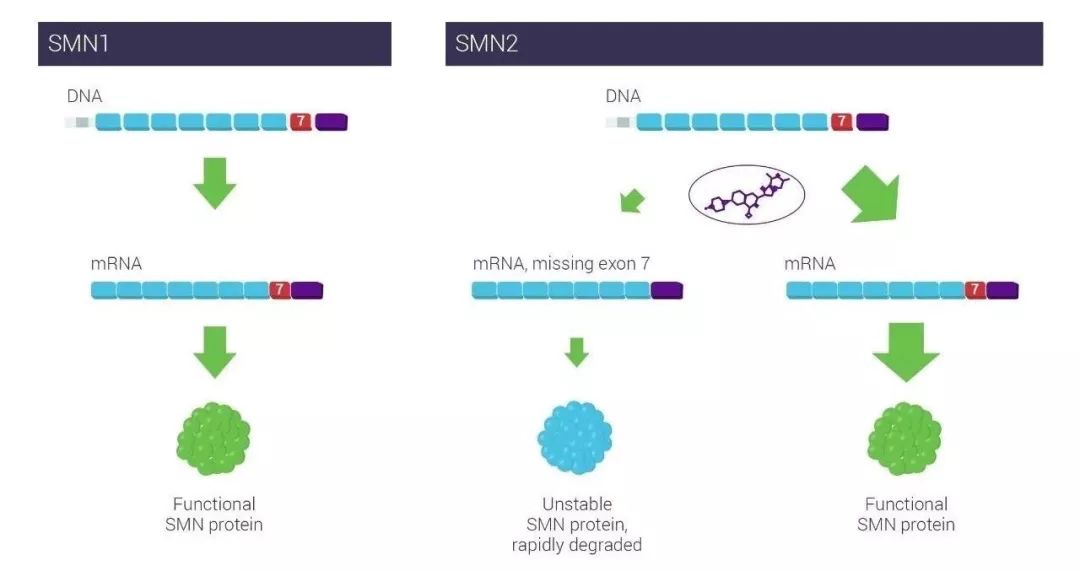
罗氏(Roche)旗下基因泰克(Genentech)公司宣布,美国FDA已经批准Evrysdi(risdiplam)上市,用于治疗年龄为2个月以上婴幼儿和成人脊髓性肌萎缩症(SMA)患者。这是首款获得FDA批准治疗SMA的口服疗法,它不但可以治疗病情最为严重的SMA婴幼儿患者,还获批治疗症状相对较轻的青少年和成人患者。Evrysdi是一款通过调节SMN2基因的mRNA剪接过程,提高运动神经元生存蛋白(SMN)水平的小分子药物。
Evrysdi由基因泰克与PTC Therapeutics公司联合开发。人体中携带的SMN2基因虽然也能够表达SMN蛋白,但是由于mRNA剪接错误,导致正常SMN蛋白表达水平很低,无法弥补SMN1基因突变导致的SMN蛋白缺失。Evrysdi通过调节SMN2基因mRNA的剪接,提高能够表达正常SMN蛋白的mRNA水平,从而缓解SMA患者的症状。Evrysdi是一种液体配方的药物,可以在家中口服或者通过饲管以液体形式给药。
▲Evrysdi通过调节SMN2 RNA剪接提高SMN蛋白水平(图片来源:PTC Therapeutics公司官网)
这一批准是基于FIREFISH和SUNFISH两项关键性临床试验的结果。在治疗2-7个月大的婴儿型SMA患者的FIREFISH研究中,41%接受治疗剂量治疗的婴儿可以在无辅助情况下维持坐姿5秒以上,这是未接受治疗婴儿无法达到的发育里程碑。此外,81%的婴儿在接受治疗23个月后无需接受永久性通气就能够生存,与疾病自然发展史相比是非常显著的改善。
SUNFISH临床试验包括180名年龄在2-25岁的晚发型SMA患者。使用名为MFM32的运动功能评估检测,接受Evrysdi治疗的患者1年后平均评分提高1.36,而安慰剂组患者评分下降0.19。
注:本文旨在介绍医药健康研究进展,不是治疗方案推荐。如需获得治疗方案指导,请前往正规医院就诊。
版权说明:本文来自药明康德内容团队,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台。转载授权请在「药明康德」微信公众号回复“转载”,获取转载须知。



 个人中心
个人中心
 我是园区
我是园区
 退出
退出