

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!
口服液体制剂单剂量包装用复合膜(袋)选择指南
Selective guideline of laminated film(bag)for oral liquid dose
(征求意见稿)
引 言
口服液体制剂包括口服溶液剂、口服混悬剂、口服乳剂、糖浆剂、合剂、酒剂等,常见的口服液体 制剂的包装有玻璃瓶、塑料瓶、复合硬片、复合膜包装。
口服液体制剂包装根据剂型包装规格可分为多剂量包装和单剂量包装。根据口服液体制剂特点及包 装形式要求,可选择不同组成结构的复合膜。口服液体制剂复合膜更适用于单剂量包装。单剂量包装规 格一般不大于30ml。
单剂量包装克服了多次开启后药液物理稳定性下降,水性液体易发生霉变等问题。单剂量复合膜(袋)包装具有剂量准确,制剂配方可以不使用抑菌剂,降低使用过程被污染的可能性,还有易携带、 储运方便、开启方便的特点。复合膜包装采用预先印制内标签,避免采用不干胶标签中相关添加剂对药 品的影响,减少不干胶粘合剂的迁移风险。
本指南可用于药品制剂注册申请人或药品上市许可持有人(以下简称药品上市许可持有人)在新药 研发和已上市药品包装变更选用的参考。
药品上市许可人选择复合膜包装口服液体制剂需满足临床适用性要求。口服液体制剂属于低风险品 种,根据口服液体制剂与包装的接触影响和包装发生相互作用的可能性,需开展相关研究。
本指南是在现行法规和标准体系以及当前认知水平下制订的,随着法规和标准的不断完善,以及科 学技术的不断发展,本指南相关内容也将进行适当的调整。不包括注册审批所涉及的行政事项,亦不作 为法规强制执行,应在遵循相关法规的前提下使用本指南。
1 范围
本指南提供了口服液体制剂单剂量复合膜包装的特点、常用组成结构及特点、选择原则及要求等内容。
2 名词术语
2.1 多剂量包装 Multi dose packaging
可以在不改变剩余部分药品安全性、剂量、质量、或纯度的情况下,连续取出该药品剩余部分的容 器密闭系统。
2.2 单剂量包装 Single dose packaging
盛有一定量药品打开后能立即使用,能够一次性给药的容器密闭系统。
2.3 复合膜袋包装 Laminated film(bag)
以袋的形式实现密闭系统,是单剂量口服液体制剂常用的包装形式。
2.4 风险源物质 Risk source substance
是指生态环境下不利影响的一种或多种化学的、物理的或生物的风险来源,本文特指复合膜中小分子单体、添加剂、助剂及非有意添加物质(降解产物、反应副产物、杂质、原料添加剂等),进入药液可能会影响药品有效性或患者身体安全性的物质。
2.5 风险源控制 Risk source control
复合膜、袋生产商在进行复合膜组成设计和生产时,应依据相关的法律法规选择符合食品、医药品 的粘合剂、油墨体系和用量要求,并能确保复合膜、袋产品配方的一致性和生产加工工艺的稳定性;应 当包含不仅限于包装材料组成、粘合剂、油墨、生产工艺、以及组成复合膜(袋)原辅料质量标准等研 究资料和评估报告,并对风险源物质采取有效的控制方法。
2.6 风险源识别 Risk source identification
针对复合膜的组成及供应商提供的产品,药品上市许可人通过对拟采用复合膜(袋)供应商的评估 报告进行综合分析,初步获得风险源物质种类、使用量和最大残留量,判断是否超过特定迁移限度。
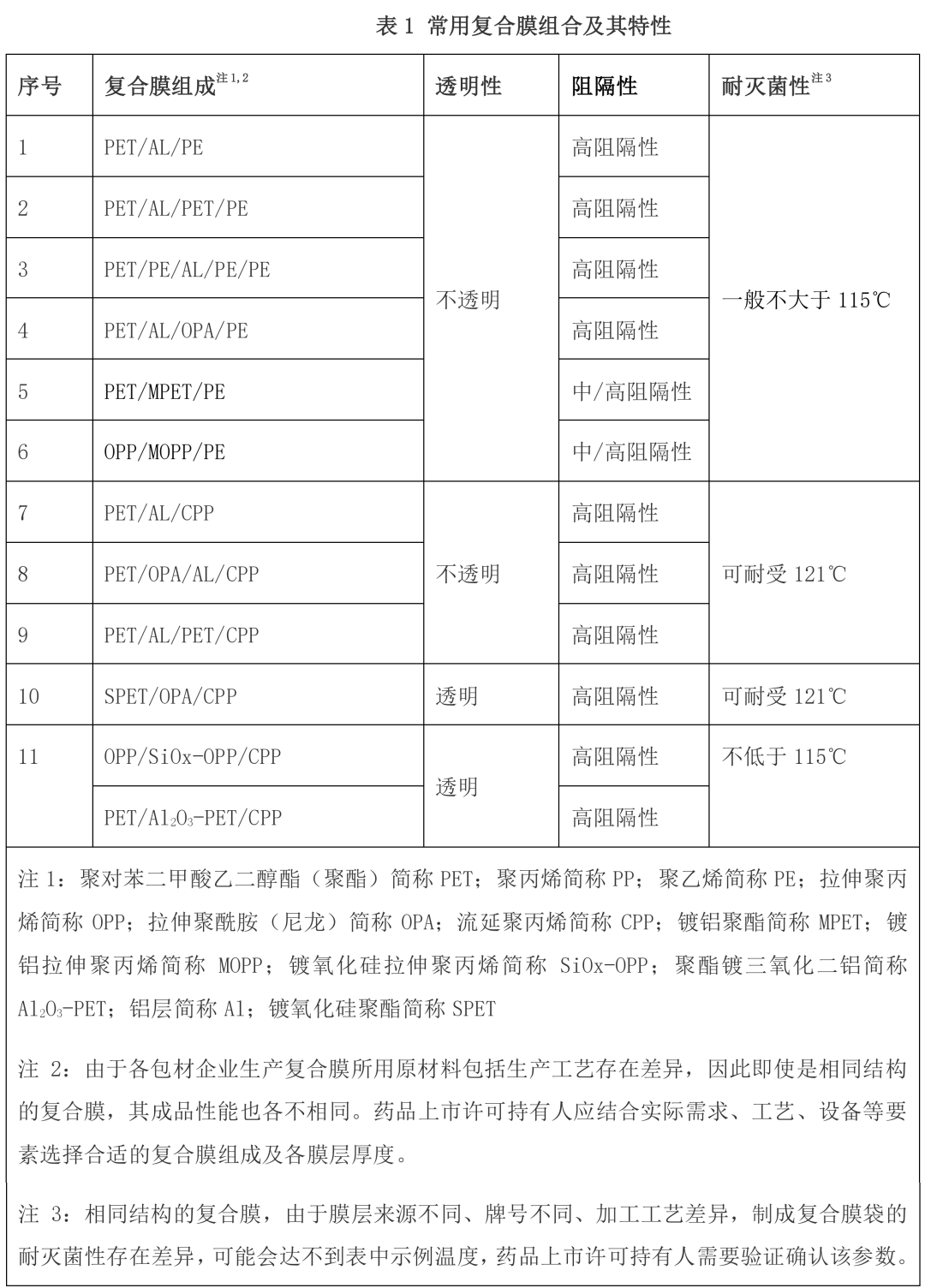
3 常用复合膜组合及其特性
口服液体单剂量制剂可采用不同组成结构的复合膜制成袋作为完整密闭系统,根据制剂特点和需求 目的选择合适的复合膜组成,不同的材质和组合方式具有不同的性能,供药品上市许可持有人根据满足 口服液体制剂产品的适用性要求进行合理选择应用。
目前常用的复合膜组合及其特性见下表 1。
表 1 常用复合膜组合及其特性
4 选用原则
单剂量口服液体制剂复合膜包装选择应从安全性、适用性、稳定性、功能性、保护性和便利性方面 综合评估(评价)。
4.1 药品适用性:口服液体制剂复合膜包装可以根据药品生产企业的药品特性、药品的工艺要求、药品 拟定的有效期、临床对该品种的需求选择不同复合膜组合。
4.2 生产环境要求:复合膜(袋)生产企业应与所包装药品生产企业的生产环境要求相适应。通过产品 生产工艺、生产环境控制以达到产品较低的生物载荷。
4.3 保护性要求:根据口服液体单剂量制剂的特点及需求目的选择合适的复合膜组成结构,如制剂要求避光,复合膜采用铝或镀铝材料作为中间层。如制剂需要高阻隔性(防止水、乙醇丢失),中间复合层采用铝层或镀氧化铝材料等高阻隔材料。
4.4 灭菌耐受性:如制剂工艺中采用热灌装、高温灭菌(消毒)可选择带有不同耐热等级的内层材料的 复合膜(袋)。
4.5 使用便利性:如临床需要开启便利性,可选择不同袋型具有撕拉、扭开或其他的开启方式。 4.6 安全性要求:见5.2
5 选用技术要求
5.1 合规性
5.1.1 复合膜生产企业应符合质量保证体系的相关要求,药品上市许可人需对其进行审计,保证符合药 用要求。
5.1.2 复合膜(袋)应符合相应的质量标准要求。
5.1.3 复合膜(袋)应按照国家药典委员会发布的《塑料和橡胶类药包材自身稳定性研究指导原则》(征 求意见稿)开展稳定性研究,确定质量稳定时限,稳定性时限应长于药品稳定性时限要求。
5.2 安全性
5.2.1 复合膜材料所用添加剂应满足药用要求,也可采用食品接触材料添加剂的法规 GB9685-2016《食品安全国家标准 食品接触材料及制品用添加剂使用标准》及增补公告的要求。使用未被该标准收载的材料和添加剂,应按照新材料进行安全性评估。
5.2.2 安全性评价应包括风险源物质的识别和鉴定,确认其使用量和最大残留量信息,判断这些风险源 物质的最大迁移量是否超过其在法规中的特定迁移限值。
5.2.3 风险源物质的识别应参考 GB9685-2016《食品安全国家标准 食品接触材料及制品用添加剂使用标准》及增补公告,研究程序、试验条件、相关检测方法可参照《口服液体制剂复合膜(袋)包装风险 源评估》,见附录2。
5.2.4 口服液体制剂使用的药包材应视情况开展相应的安全性研究。如果处方为含醇制剂应针对处方成 分进行加严条件下的提取研究,以确定风险源物质是否在可接受范围之内。若风险源物质在可接受范围 之内,经评估,有必要时酌情进行相容性研究;若风险源物质超出了可接受范围,则建议更换复合膜。
5.3 生产工艺适用性
5.3.1 应关注包装对于口服液体制剂的工艺适用性,如果口服液采用灭菌工艺,应考虑复合膜材耐灭菌 的适用性。
5.3.2 工艺符合性研究:灌装、消毒或灭菌(如适用)、包装工序应考虑设备的适用性。袋的封口应平 整、压痕或压纹清晰、无皱褶、灼化和压穿现象。袋上生产日期、生产批号、标识系统等应清晰、牢固, 打印(开口)位置应一致。封口强度、封口宽度、装量偏差等应符合工艺标准要求。
5.3.3 吸附和残留影响:应充分考虑吸附和残留均有可能影响给药剂量的准确性。
吸附影响:重点关注物理吸附,主要有内层材料吸附,如复合膜吸附药液使有效成份降低。
残留影响:药液残留与复合膜袋型设计有关,同时与容器结构或器型设计有关,如:罐装工艺过程中流 道内药液的残留,或袋型设计留有死角等;
5.3.4 干燥形式:采用不同的干燥形式对复合膜材料的挥发性物质的残留有直接的影响。选用适宜的干 燥方式的参数,如烘干温度、时间和真空度等;
5.3.5 输送方式:成品输送对袋型外观的影响;无碰撞、挤压风险;
5.3.6 储藏条件:应考虑不同地区温度、气压对复合膜产品的影响。
5.4 功能性评价
5.4.1 容器形状:一般采用条形包装。如为易撕设计,应考虑易撕线尺寸范围(适用时)、易撕部位及 撕拉强度;
5.4.2 耐压性:耐压性可以采用跌落方式进行。跌落试验可参考标准 GB/T 17313-2009《袋成型-充填-
封口机通用技术条件》
5.4.3 内标签完整性、清晰度评价:内标签内容应符合相关的法规要求,并符合完整、清晰的要求,需
考虑在线打印标签过程中,打码对卷膜阻隔性的影响;灭菌对打码字迹清晰度的影响;卷膜标签字迹清 晰度;非破坏性手段是否能够擦拭打码字迹等。
5.5 保护性
如密封性、热合性能、阻隔性能等,应考虑复合膜材和所用药品在不同地区储存运输过程中震动、 温度、气压等对成品的影响。
5.6 便利性
应具有便利的开启性能。开启性能如开启方式、开启力、开口质量应符合开启后无明显拉丝、毛边; 开启过程无飞溅、滴漏等。需要时应考虑老年和儿童用药的友好方式。
5.7 质量标准建议内容
5.7.1 项目设置
复合膜、袋质量标准检测项目的设置应能够反映产品质量的变化情况,控制项目一般包括但不限于 外观、鉴别、膜水蒸气透过量、袋水蒸气透过量(如适用)、溶剂残留量、复合膜结构形式、微生物限 度、异常毒性、功能性、保护性和便利性、膜(袋)风险源物质(如适用)、如:芳香族伯胺,环氧硅 烷偶联剂等项目。
5.7.2 袋溶出物试验
所选用的复合膜(袋)要与口服液制剂的配方及有关工艺、包装规格相适应,溶出物试验检验项目 应包括不仅限于“澄清度、吸光度、pH变化值、易氧化物、不挥发物、重金属”。质量标准包括检测项目、分析方法和可接受标准。符合标准是指按照拟定的分析方法检测,结果符合可接受标准。复合膜 参照袋溶出物试验项目。
5.7.3 稳定性研究
稳定性研究按照《中国药典》2020 年版四部 9001《原料药与制剂稳定性试验指导原则》及 CDE 相关稳定性研究技术指导原则的要求进行。
稳定性试验用样品应选择完整包装并与上市或商业化生产产品相同或相似。
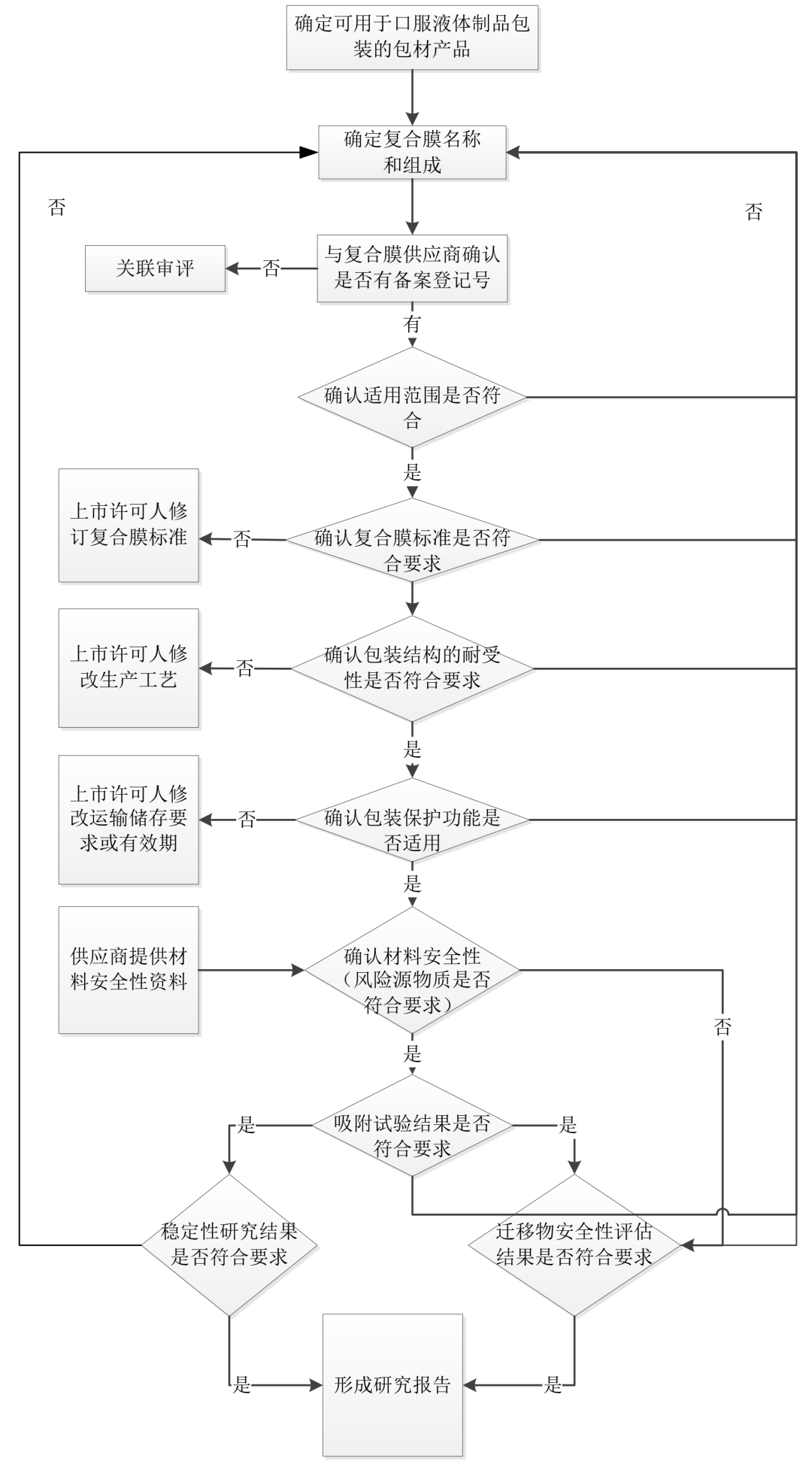
附录 1 单剂量口服液体制剂复合膜包装选择决策树
附录 2 口服液体制剂复合膜(袋)包装风险源评估
1 风险源物质来源
口服液体制剂单剂量复合膜(袋)风险源物质主要来自:组成复合膜的原材料(如:薄膜、粘合剂、 油墨)添加剂,和生产加工用助剂。按照表 2《常见风险源物质名称和限度》示例,材料和组成不同、生产工艺不同迁移物的种类和数量也不同。粘合剂、油墨、材料中涉及的小分子单体、添加剂、助剂及 非有意添加物质(如:反应副产物、降解产物、杂质等)是主要风险来源,这些风险源物质在不同的复 合膜(袋)供应商、不同材料之间存在差异,所以需要依据其来源分别进行风险源物质识别。
组成复合膜(袋)的材质应分别符合食品安全国家标准的相关规定,如:GB 4806.1-2016《食品安全国家标准 食品接触材料及制品通用安全要求》、GB 4806.7-2016 《食品安全国家标准 食品接触用塑料材料及制品》、GB 4806.9-2016 《食品安全国家标准 食品接触用金属材料及制品》等。对于复合膜、油墨和胶水的标准目前还是征求意见稿,请及时关注进展。
2 风险源物质迁移试验样品制备
针对产品中的风险源物质,一般通过迁移试验的方法对所迁移物进行鉴别和评估。口服液体制剂产 品属于低风险类型,可以采用有关的食品安全国家标准提供的方法开展迁移物识别及评估。采用法定的 试验方法,即可以保证科学、合理和统一,又可以减少非标的试验方法和数量,并且试验结果可以相互 比较,以利于选择使用更合适的复合膜结构、配方和供应商。
2.1 试验样品预处理按照 GB5009.156-2016《食品安全国家标准 食品接触材料及制品迁移试验预处理方法通则》和 GB 31604.1-2015《食品安全国家标准 食品接触材料及制品迁移试验通则》要求进行。若以上标准不适用,制剂企业可自行确定合理的方法或参考其他更合适的预处理方案。
2.1.1 样品预处理:复合膜(袋)应洁净,无污染。通常应与生产使用保持一致。
2.1.2 试验样品制备:
a. 复合膜 随机取筒膜,弃去前 0.5 米,按要求截取适宜的尺寸,记录接触面积(S)。
b. 复合膜袋 随机取需要的袋数,按制剂拟接触的影响较大的面积:体积比的复合膜袋,计算内表面积
(S)。
2.1.3 浸提液选择:模拟药品实际情况,可有针对性地采用如:4%乙酸溶液、20%乙醇溶液、50%乙醇溶液、口服液体制剂载体溶液或不含药物的空白制剂(适用时))或药品溶液。
2.1.4 浸提液制备:
a. 一般采用试样接触面积(S)与浸提液体积(V)之比 S/V 为 6 dm2:1L;浸提液应尽可能严苛于或接近于实际情况,如果与实际情况不相符,试验结果需折算到实际 S/V 的情况进行。
b. 药品上市许可人可采用接触影响较大的包装规格的药品,并进行接触影响计算。
2.1.5 采用灌装法:
a. 复合膜采用迁移测试池法,按试验装置操作方法放置截取适宜的尺寸,加规定体积的浸提液。
b. 复合膜袋采用制袋法,取复合膜袋加规定体积的浸提液采取物理方式排空,将试验样品进行封口,放 置试验样品,应保证样品与浸提液完全接触。
2.2 特定迁移试验条件按照 GB31604.1-2015《食品安全国家标准 食品接触材料及制品迁移试验通则》要求进行。
2.2.1 复合膜加速试验条件:对在常温下长期储存的制剂,一般推荐采用的试验条件,如:60℃,10 天;对需经高温灭菌的制剂,建议采用较严苛的条件,如:121℃,0.5 小时。也可以结合制剂实际情况选择更合适的加速试验条件。
2.2.2 复合膜(袋)及已灌装口服液体制剂的复合膜袋样品或稳定性试验样品采用加速试验条件,如: 60℃,10 天,放冷。或者按照原料药物与制剂稳定性试验指导原则(《中国药典》2020 年版四部通则9001 项下)进行。
2.2.3 预期复合膜(袋)需要经过灭菌处理则取袋加规定体积的浸提液采取物理方式排空,将加装浸泡溶剂后的复合膜袋进行封口,采用条件如:121℃,0.5h,放冷后再采用加速试验条件如:60℃,10 天,放冷(采用已灌装口服液体制剂的复合膜袋或使用稳定性试验样品可免除该步骤)。或者按照原料药与 制剂稳定性试验指导原则(《中国药典》2020 年版四部通则 9001 项下)进行。
3 风险源物质鉴别
参考常用不同复合膜组合(表 1)常见的风险源物质名称和限度及按照 GB9685-2016《食品安全国家标准 食品接触材料及制品用添加剂使用标准》及增补公告等,选择适合的专属性检测方法。
通常的检测设备包括:气相色谱-质谱(GC-MS)、液相色谱-质谱(LC-MS)、离子色谱(IC)、电感耦合等离子体发射光谱(ICP)、原子吸收光谱法(AAS)等。
按照限度要求,结合药品日给药剂量,选择灵敏度符合要求的分析方法。按照分析方法验证指导原 则(《中国药典》2020年版四部 9101)进行专属性、准确度、精密度、线性、检测限和定量限等进行方法学验证。
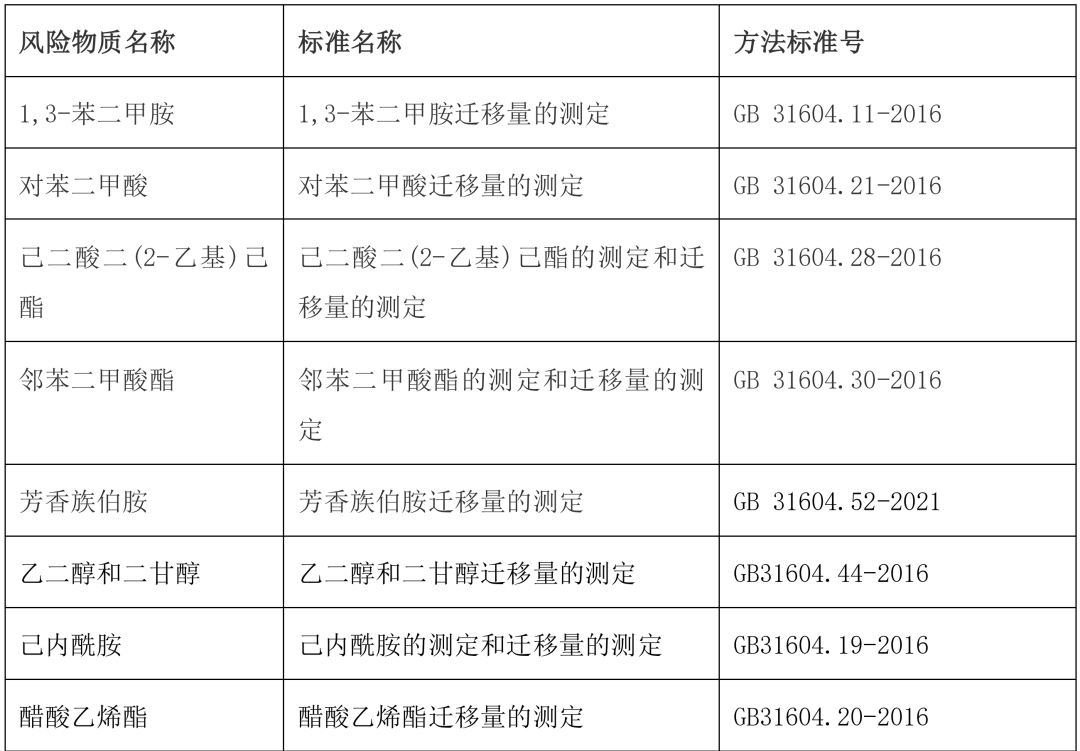
采用食品安全国家标准相关迁移物测定方法详见表 3《风险源物质可以采纳的分析方法》,并参照分析方法确认指导原则(《中国药典》2020 年版四部 9099)进行方法学确认。
本指导原则提供了单剂量口服液体制剂复合膜常见风险源物质名称和限度,见表 2。(注:风险源物质限度要求可参考 GB9685-2016《食品安全国家标准 食品接触材料及制品用添加剂使用标准》及其增补公告)
4 风险源物质限度确认
根据特定迁移试验获得的风险源物质信息,进行必要的化合物归属或结构鉴定并分析汇总其含量。 风险源物质可以采用表 3 的分析方法,并根据试验结果,按照 GB9685-2016《食品安全国家标准 食品接触材料及制品用添加剂使用标准》及增补公告判断是否符合规定。
表 2 常见风险源物质名称和限度
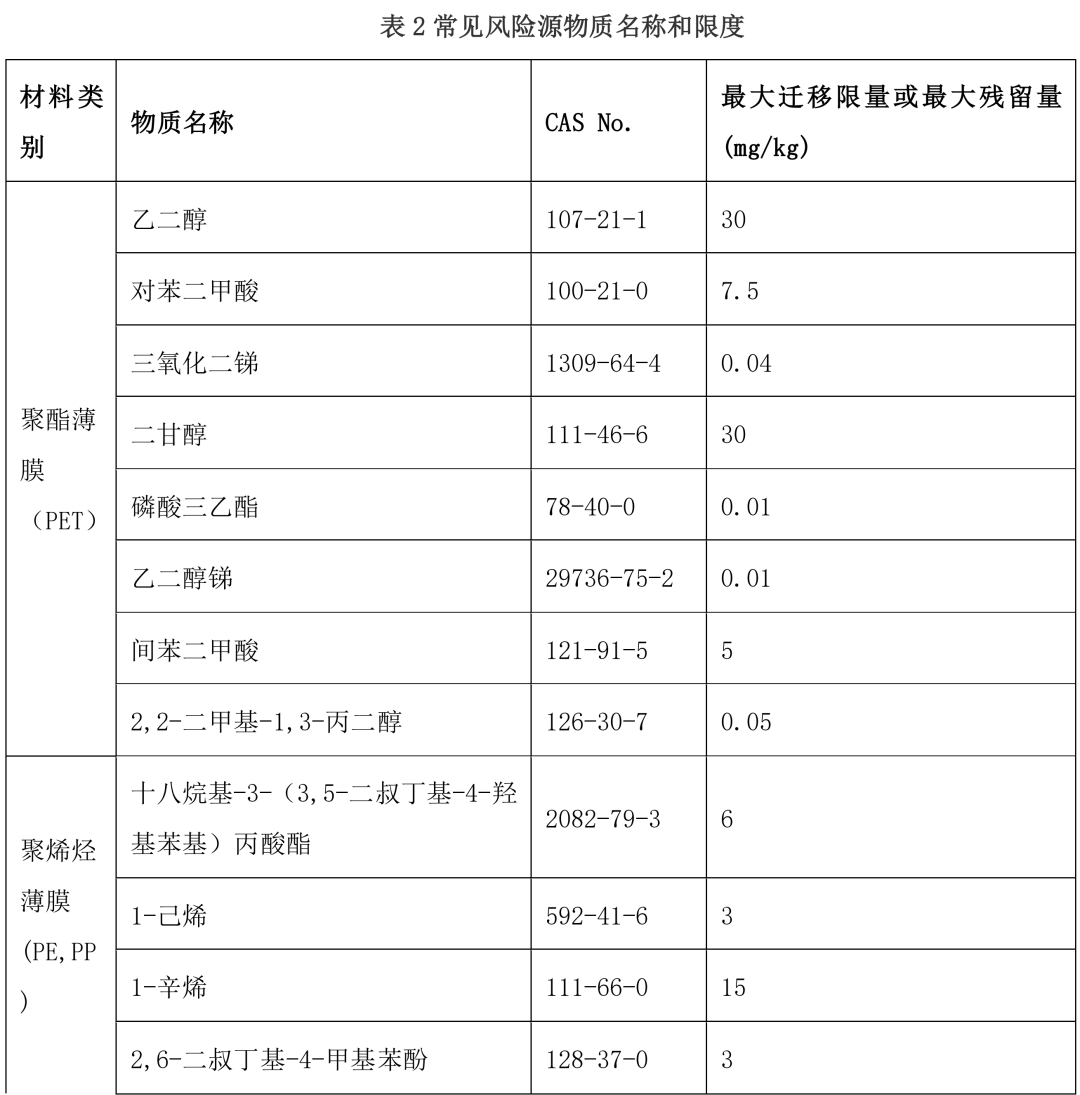
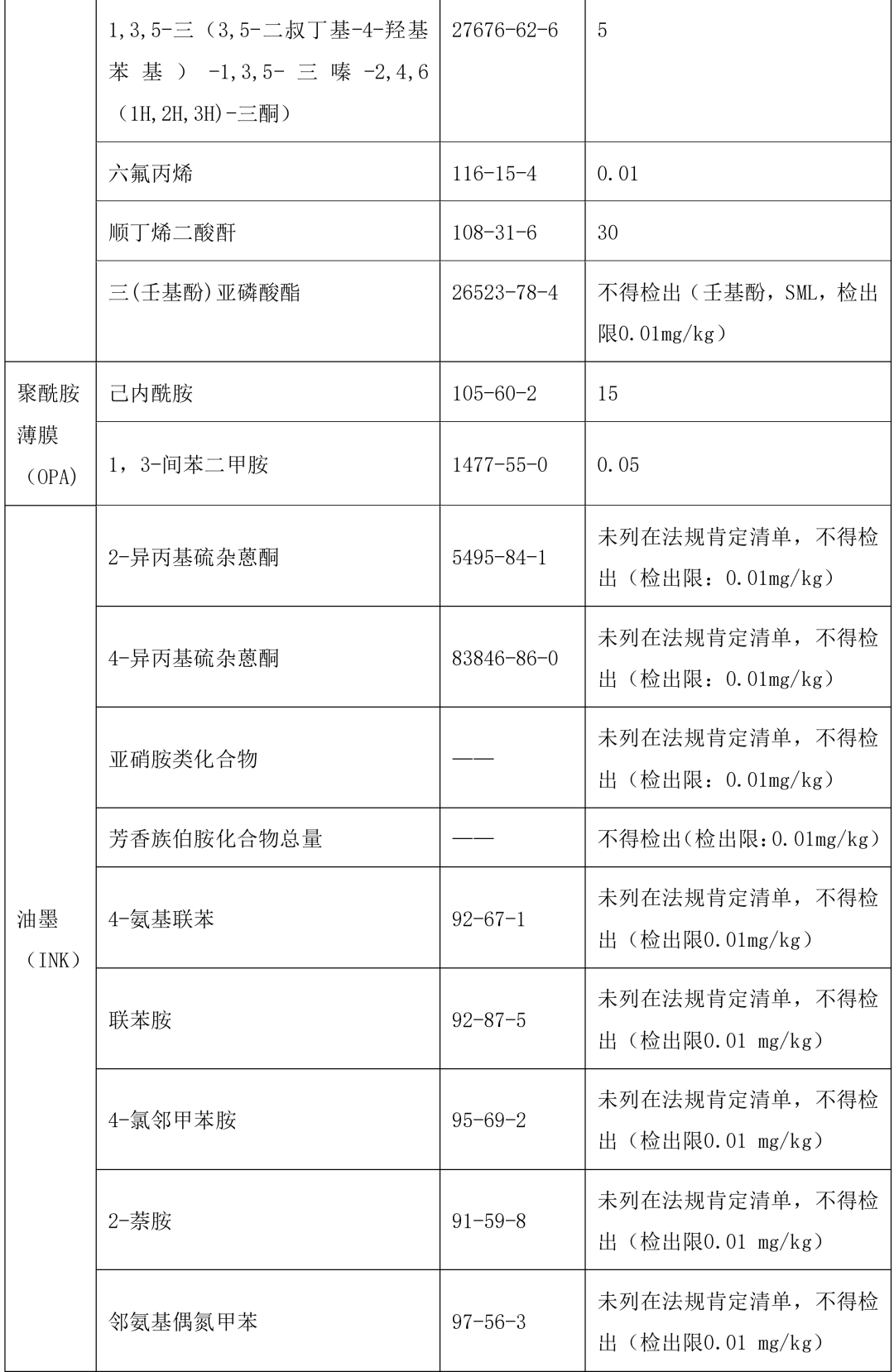

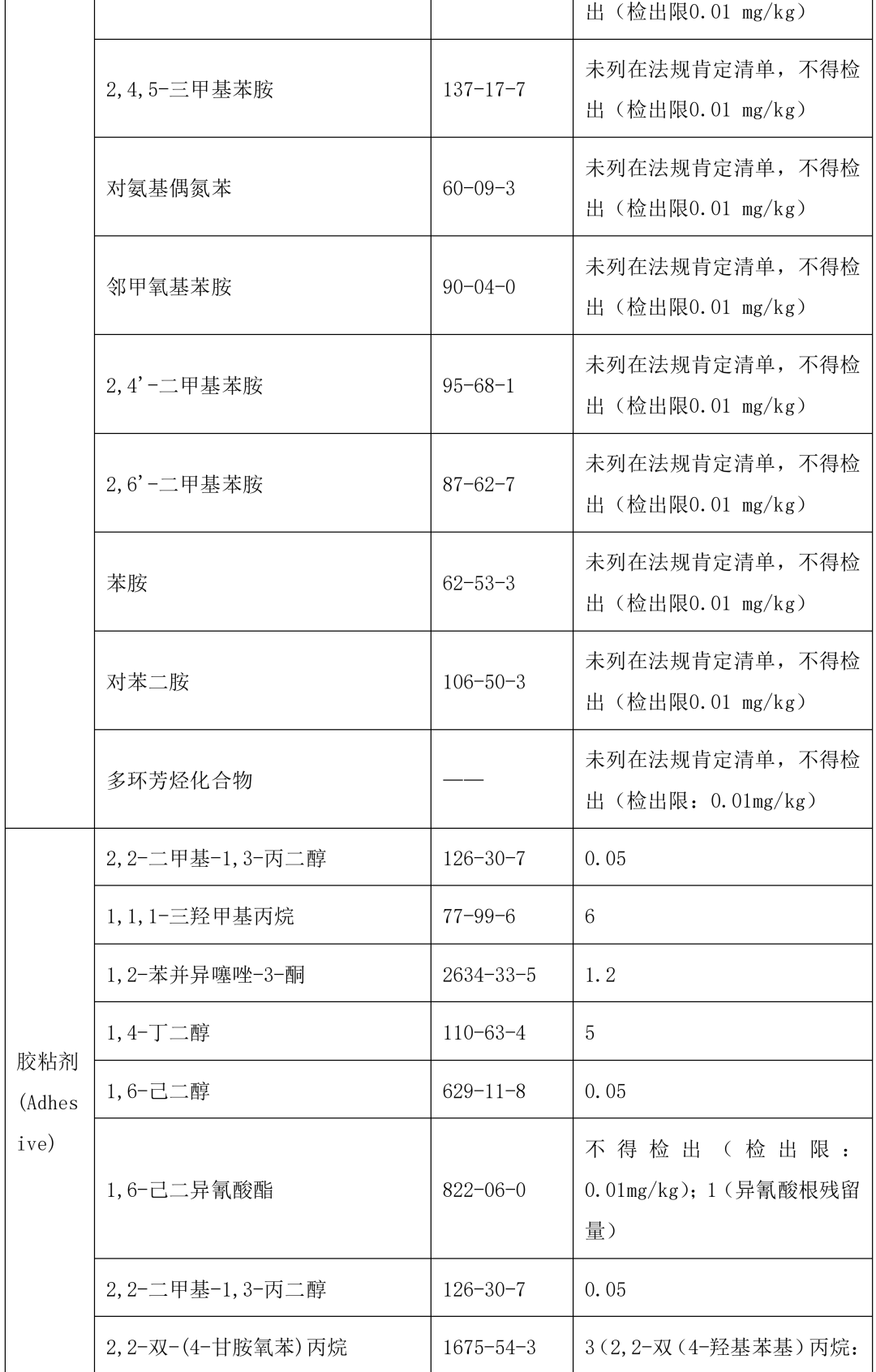
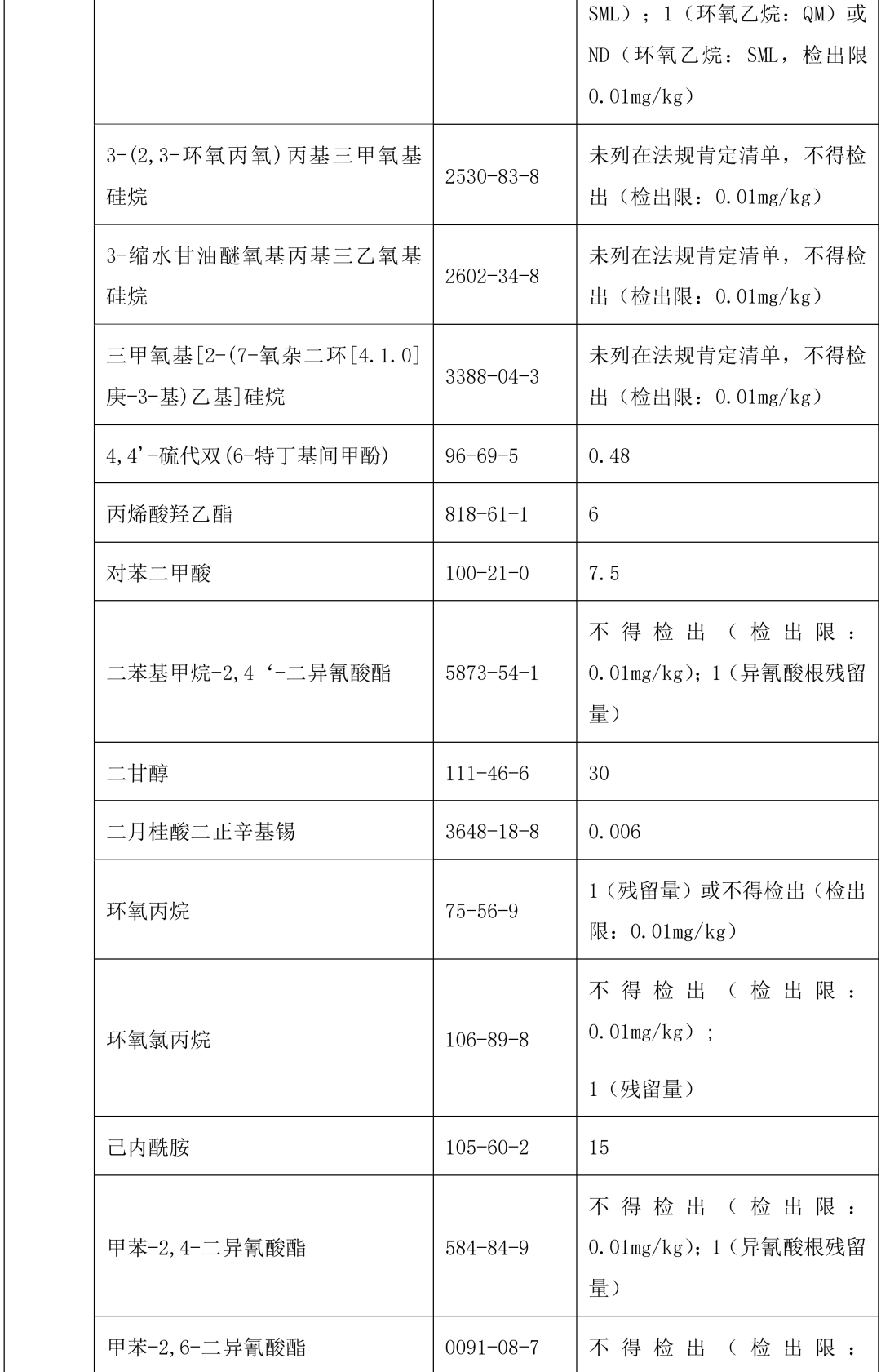
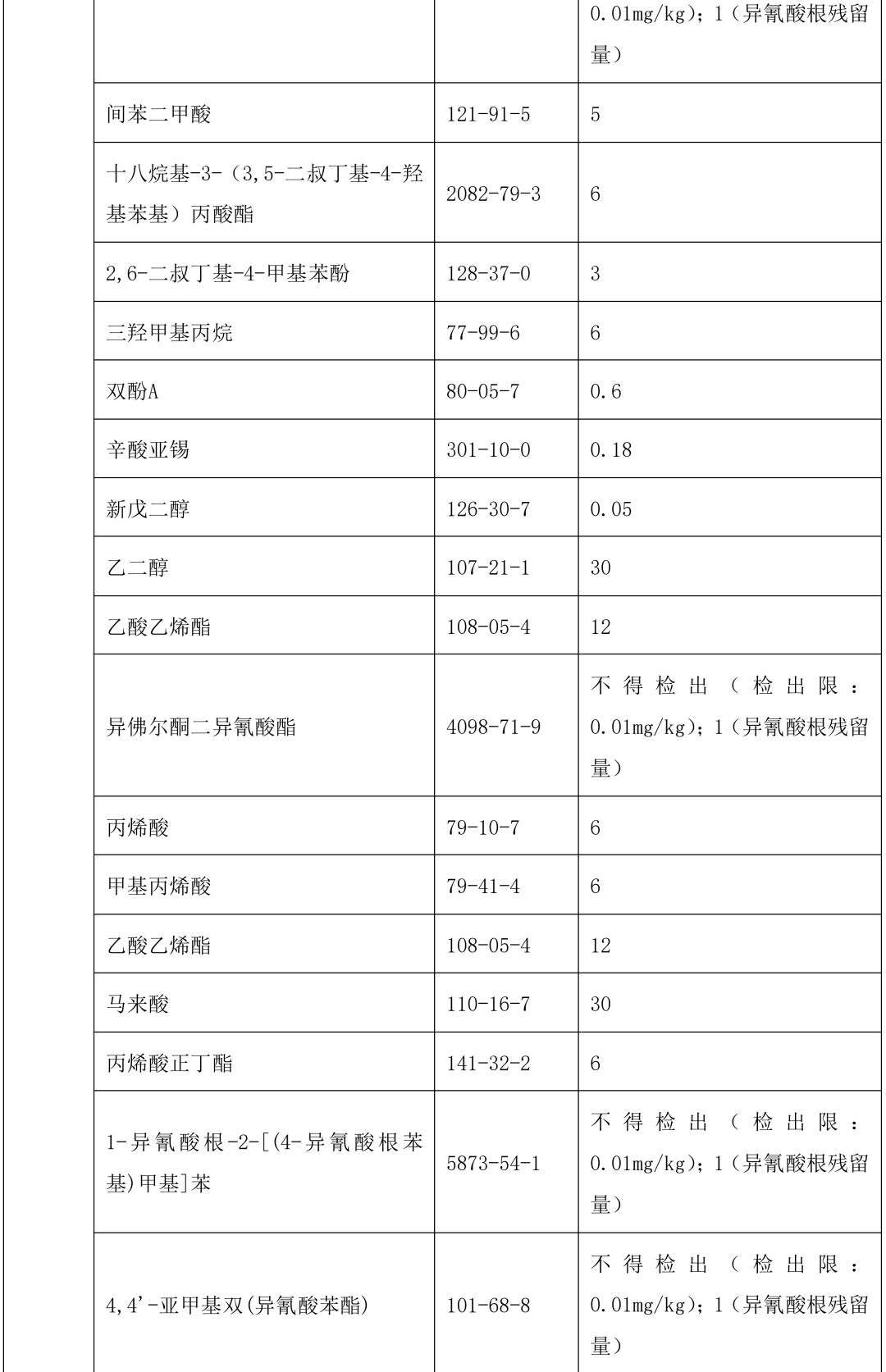
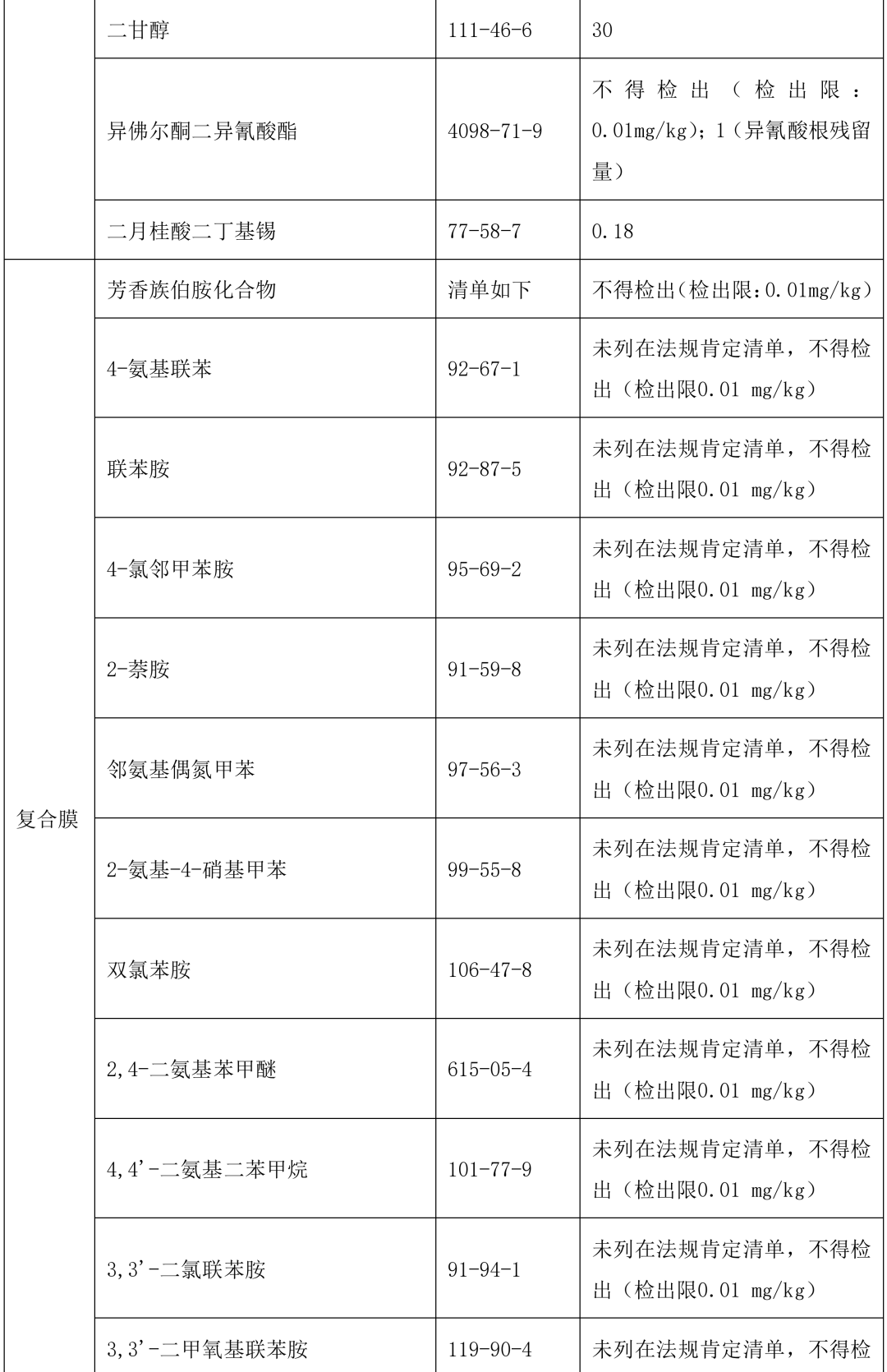
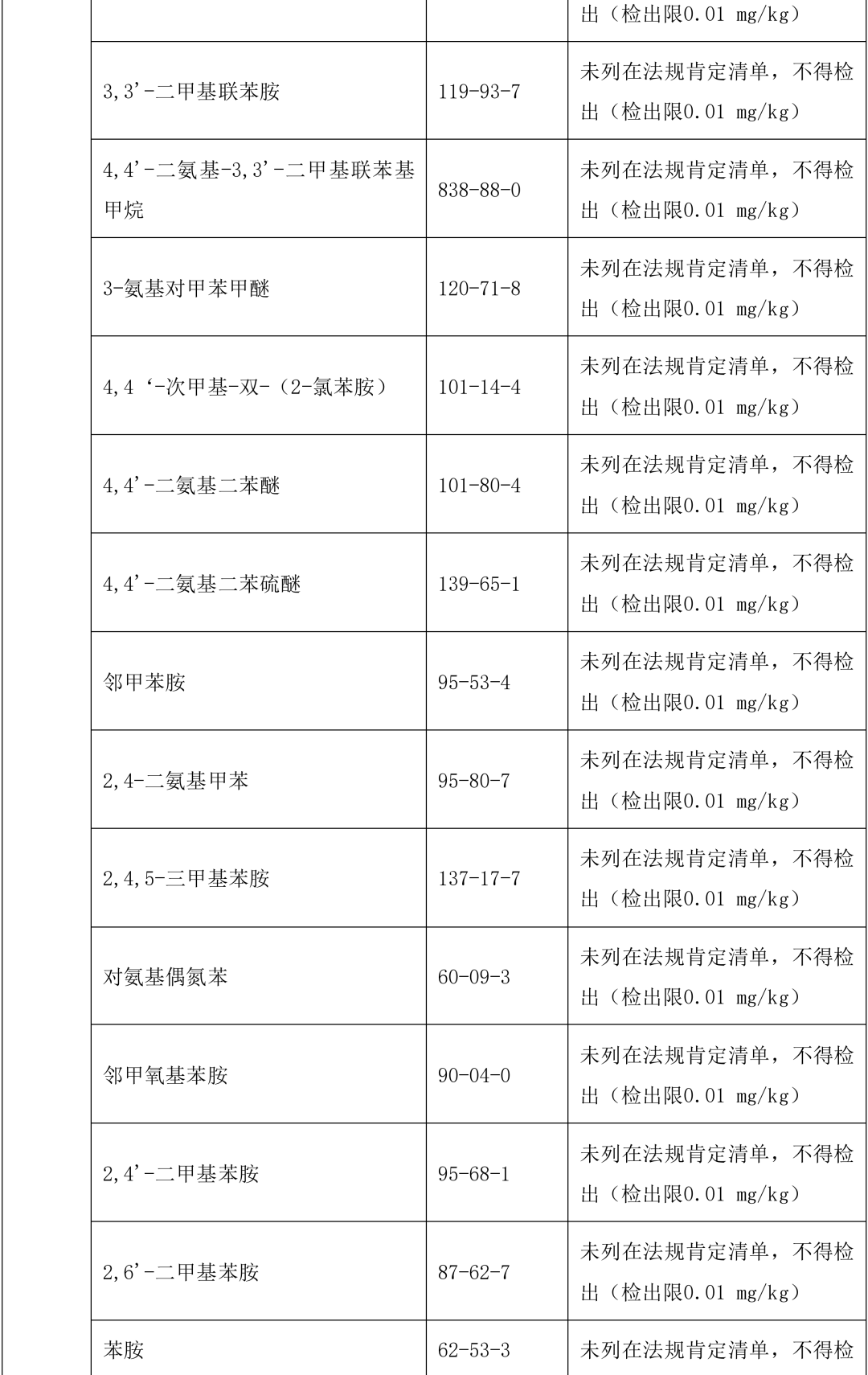
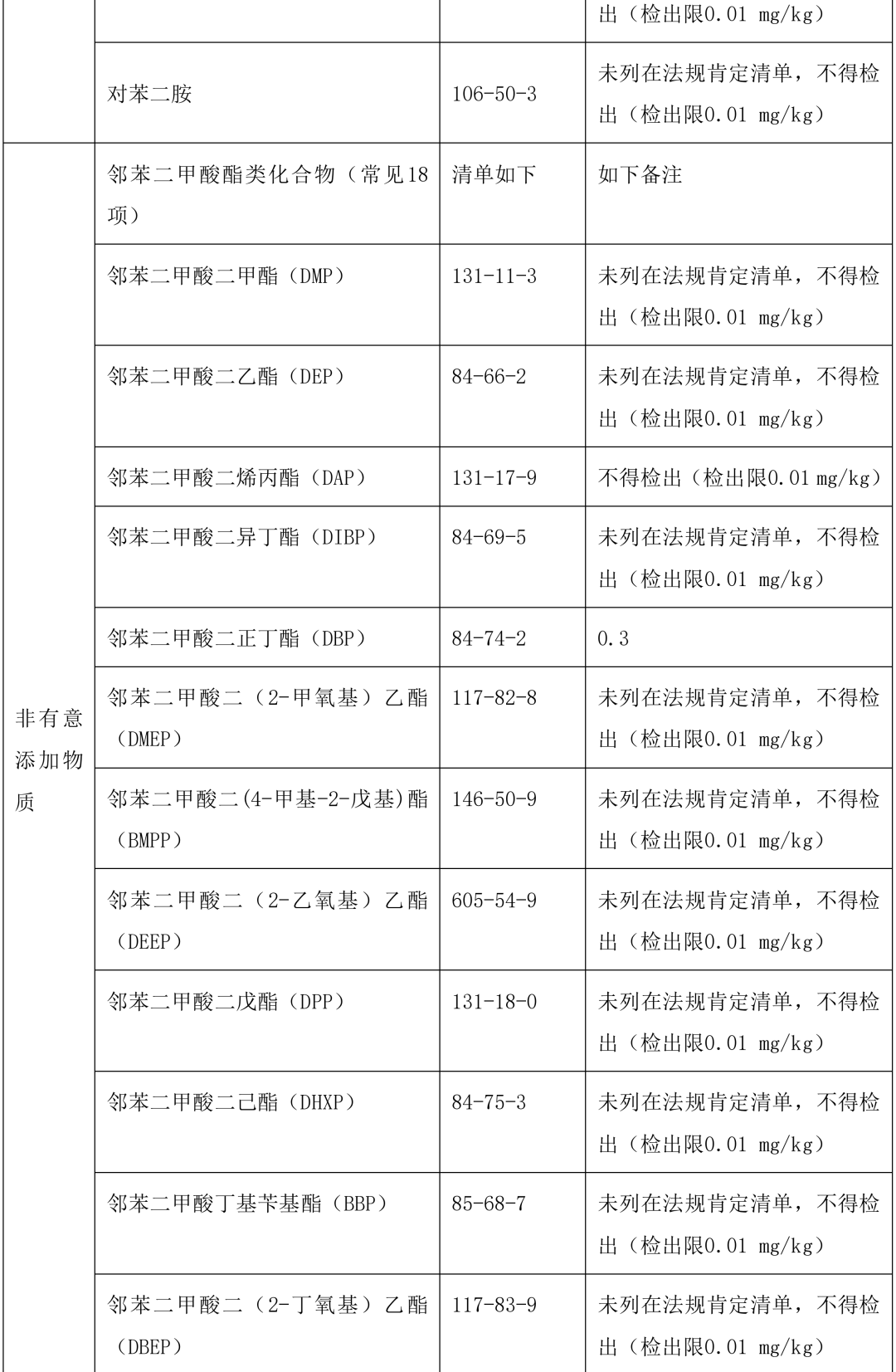
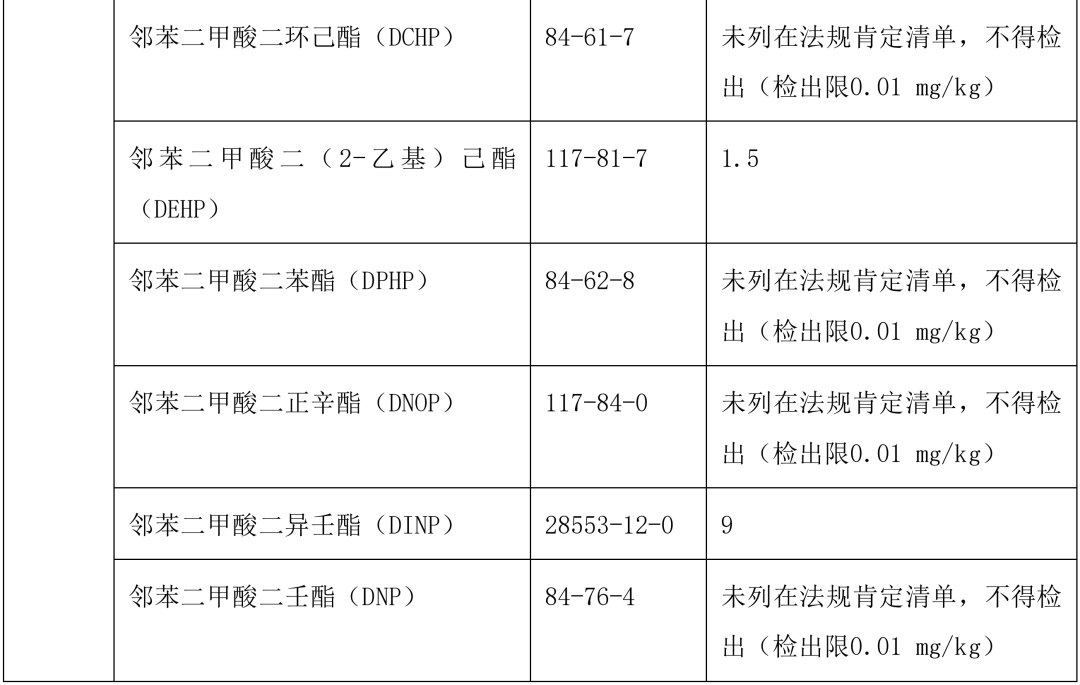
表 3 风险源物质可以参照的分析方法






公众号
GMP办公室

专业的GMP合规性研究组织
国内外(FDA、EMA、MHRA、NMPA、WHO、PIC/S等)GMP法规解读;
国内外制药行业GMP监管动态;
GMP技术指南(ISPE、PDA、ISO、ASTM等)分享








