

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

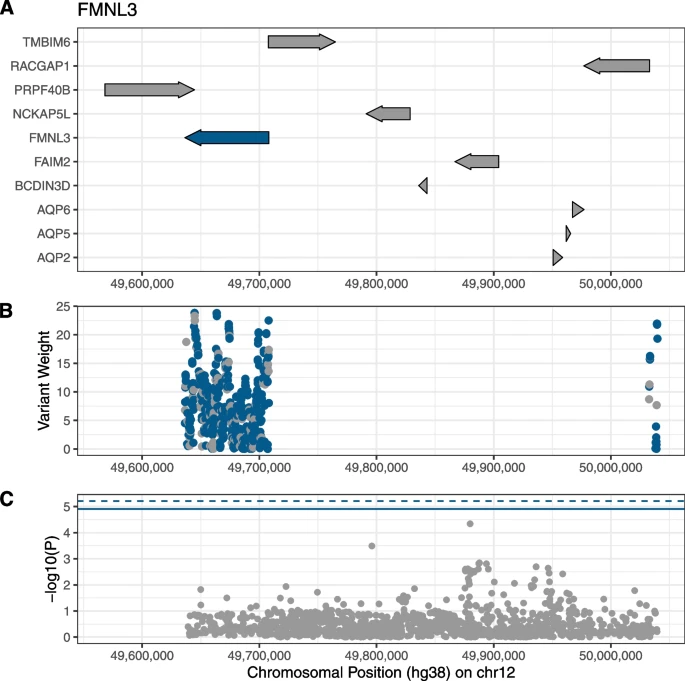
乳腺癌是全球女性最常见的癌症。乳腺癌的遗传性大约在13%~30%,并且很大程度上遵循复杂的遗传结构。识别乳腺癌中的疾病易感基因将有助于了解病理进程和发现新的临床生物标志物或药物靶点。但在全基因组关联研究(GWAS)中发现的单标志物关联与靶基因的关联仍未明确,阻碍了对疾病发生机制进一步的理解。



目前已有500+行业精英加入基因俱乐部
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()


乳腺癌是全球女性最常见的癌症。乳腺癌的遗传性大约在13%~30%,并且很大程度上遵循复杂的遗传结构。识别乳腺癌中的疾病易感基因将有助于了解病理进程和发现新的临床生物标志物或药物靶点。但在全基因组关联研究(GWAS)中发现的单标志物关联与靶基因的关联仍未明确,阻碍了对疾病发生机制进一步的理解。
目前已有500+行业精英加入基因俱乐部
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()






