信达 PI3Kδ 抑制剂报上市,诺华「司库奇尤单抗」新适应症获批… | Insight 创新药周报
收藏
关键词:
单抗上市新药创新药周报抑制剂制剂适应症获批诺华信达药
资讯来源:Insight数据库 + 订阅账号
发布时间:
2022-10-31
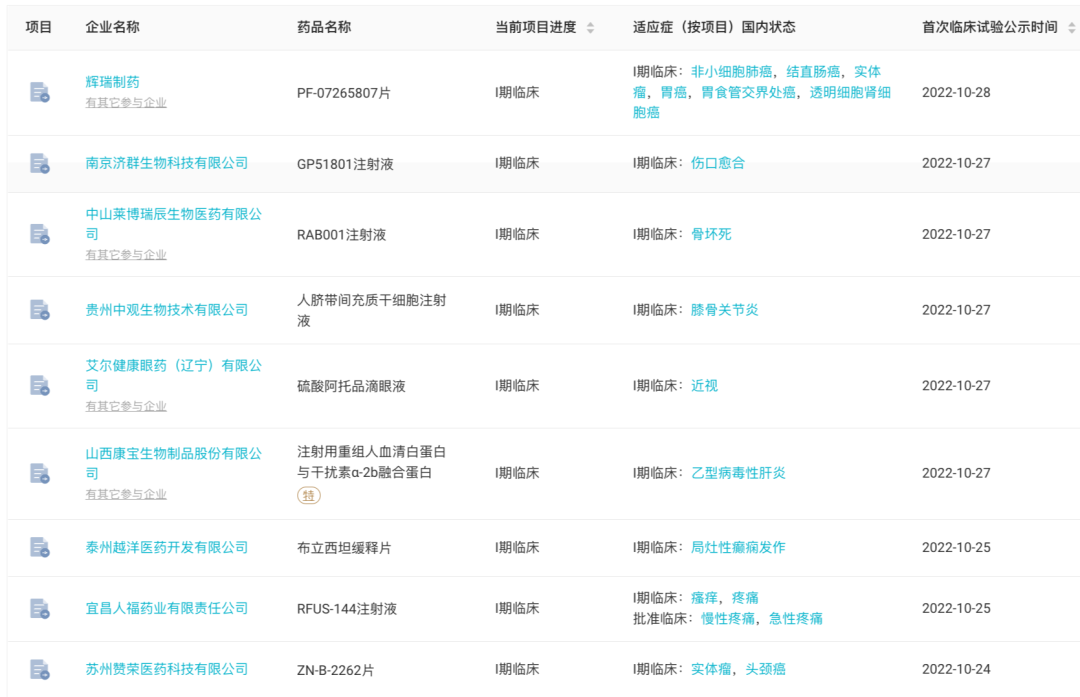
据Insight 数据库 统计,上周(10 月 24 日 - 10 月 30 日)全球共有 45 款创新药(含改良新)研发进度推进到了新阶段,其中 8 款首次申报临床,14 款首次获批临床。
下面,Insight 将分别摘取国内外部分重点项目做介绍。
国内方面,本周共有 31 款创新药(含改良新)研发进度推进到了新的阶段
,其中,申报临床 8 款,获批临床 9 款,首次公示临床试验的有 9 款。
来自:Insight 数据库网页版(http://db.dxy.cn/v5/home/)
1、信达生物:PI3Kδ 抑制剂申报上市,拟优先审评
10 月 25 日,CDE 官网显示,信达生物的 PI3Kδ 抑制剂 Parsaclisib 已经递交上市申请,并拟纳入优先审评,用于既往接受过至少两种系统性治疗的复发或难治性滤泡性淋巴瘤成人患者。
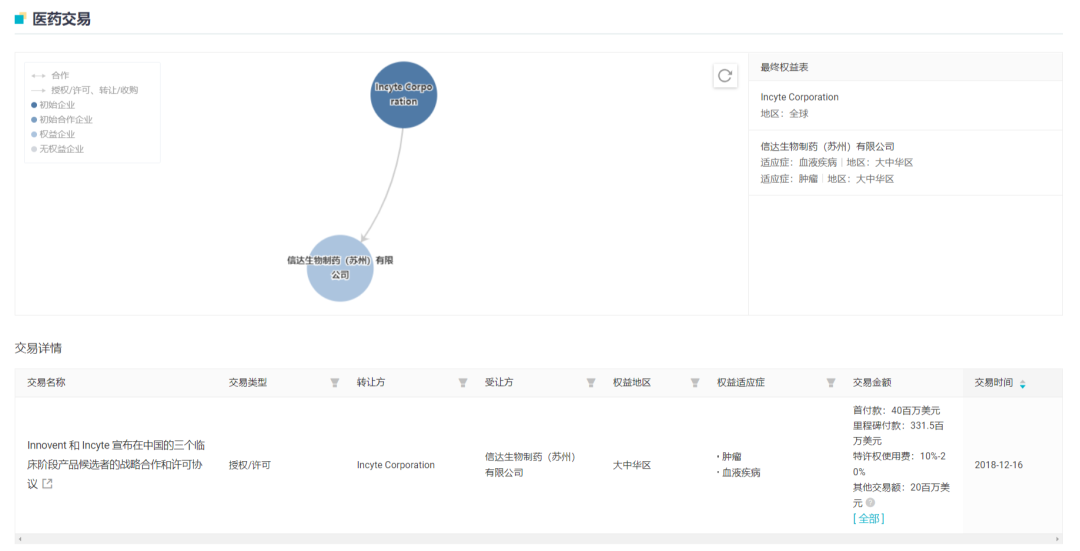
Parsaclisib 来源于信达 2018 年度与 Incyte 公司的合作。在该项合作中,信达生物获得了 3 款抗肿瘤药物在中国内地及港澳台地区的临床开发和商业化权益,而 Incyte 公司则获得信达生物的 4000 万美元首付款、首次递交 IND 申请后的第二笔 2000 万美元付款、潜在开发及监管里程碑款最高 1.29 亿美元和潜在商业里程碑 2.025 亿美元。
这 3 款药物分别为:itacitinib(JAK1 抑制剂)、pemigatinib(FGFR 抑制剂)和 parsaclisib(PI3Kδ 抑制剂)。在国内,佩米替尼(pemigatinib)已经于今年 4 月获批上市,如今 parsaclisib 紧随其后也递交了上市申请,itacitinib 则处于 1/2 期临床中。
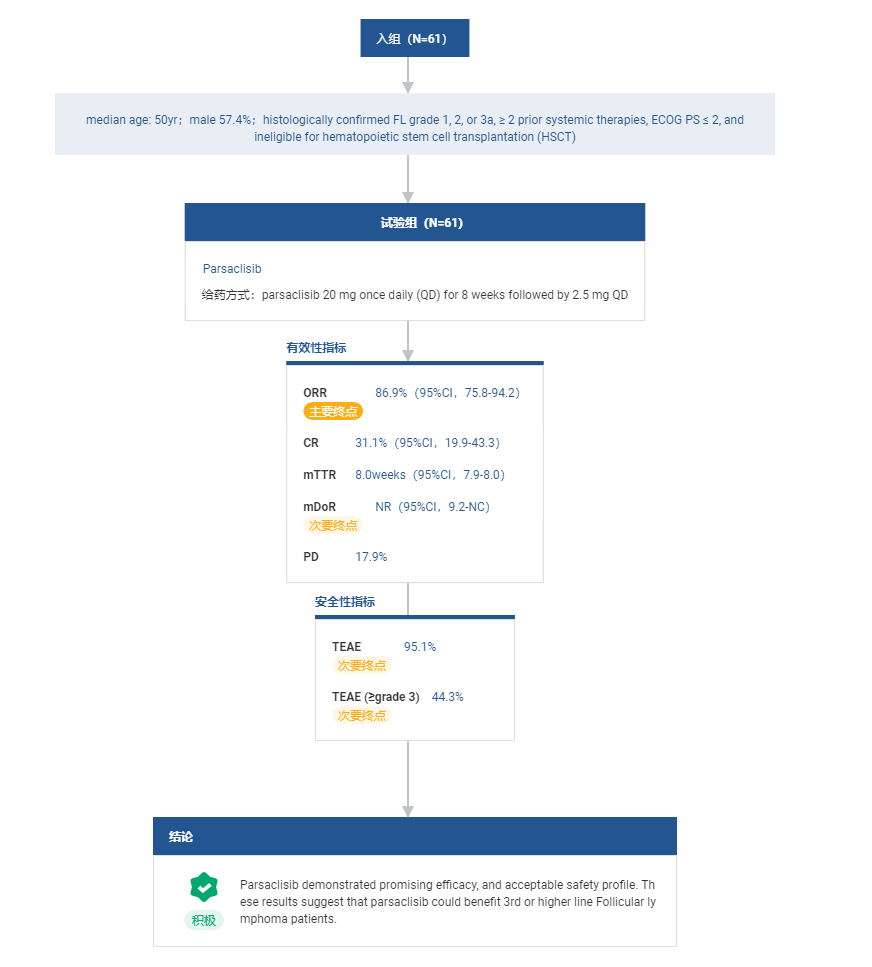
信达生物在 2022 ASCO 会议上发布了 Parsaclisib 在中国人群中的数据:在滤泡性淋巴瘤患者(N=61)中 ORR 达到 86.9%(95%CI,75.8-94.2),其中 31.1%(95%CI,19.9-43.3)达到 CR:
Parsaclisib 中国人群数据 @2022ASCO
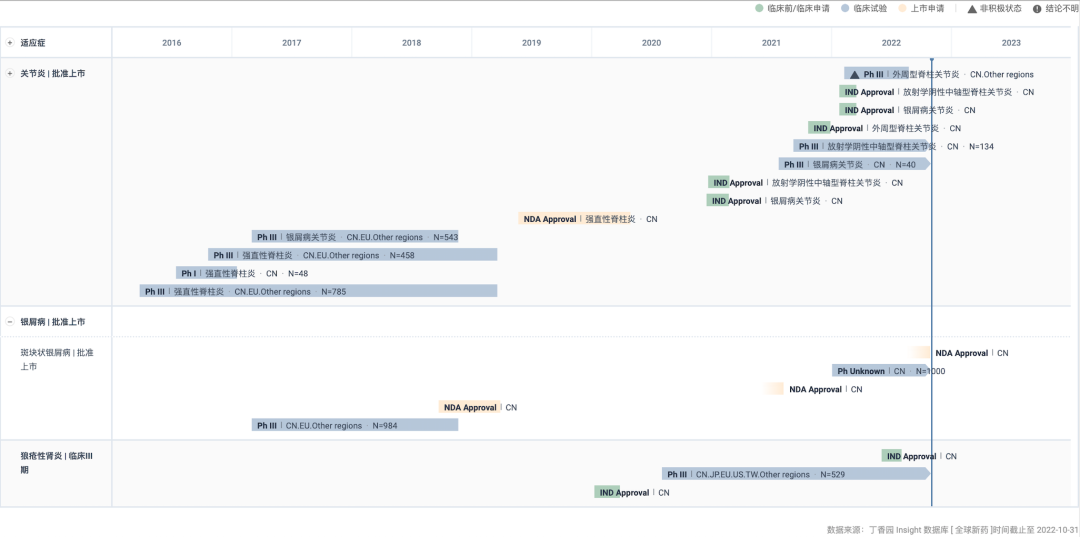
国内来看,Parsaclisib 是第 3 款申报上市的 PI3Kδ 抑制剂。今年 3 月,石药引进的 PI3Kγ/δ 抑制剂度恩西布已经获批用于滤泡性淋巴瘤;恒瑞和璎黎合作的林普利司自去年 5 月申报上市,也有望在年内获得批准。
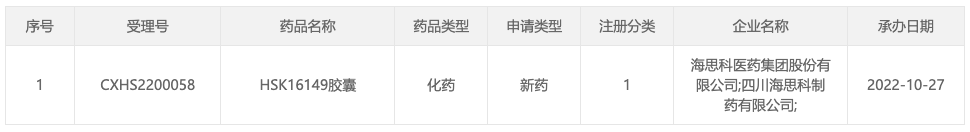
2、海思科:1 类镇痛新药 HSK16149 申报上市
10 月 27 日,据 CDE 官网显示,海思科 1 类镇痛新药 HSK16149 申报上市
(受理号:CXHS2200058)。根据临
床研究进展来看,推测此次获批适应症应为糖尿病周围神经病变。
HSK16149 胶囊是由海思科开发的具有自主知识产权的拟用于治疗糖尿病周围神经痛、带状疱疹后神经痛、纤维肌痛的 1 类新药。
临床前研究表明 HSK16149 具有强效镇痛、长效镇痛、中枢副作用小等特点,有望替代普瑞巴林、加巴喷丁,具有成为慢性神经性疼痛首选用药的潜力。
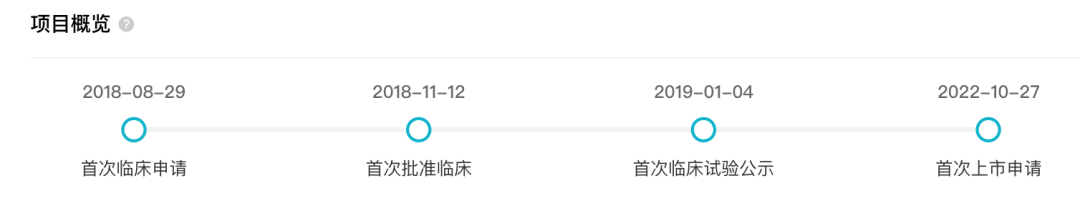
据 Insight 数据库显示,HSK16149 于 2018 年 11 月首次获批临床,次年 1 月首次公示临床实验,并于本周申报上市。
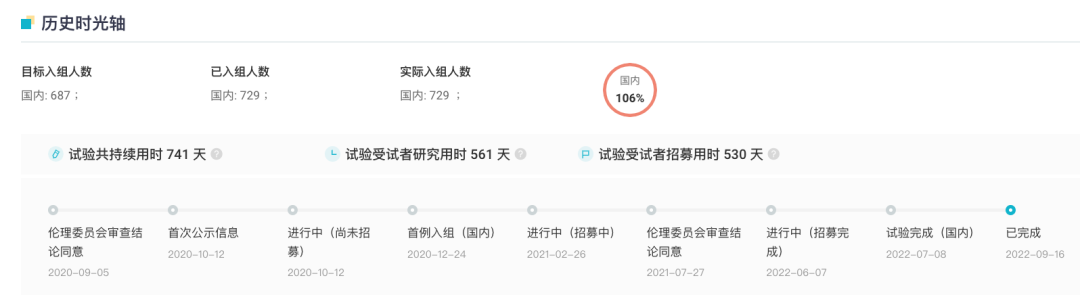
针对糖尿病周围神经痛(DPNP),据 Insight 数据库显示,HSK16149 于 2020 年 1 月首次公示一项多中心、随机、双盲、安慰剂和普瑞巴林胶囊对照的 II/III 期临床试验(登记号:
CTR20202015
),旨在评估 HSK16149 胶囊在中国糖尿病周围神经痛患者中的有效性和安全性。该试验共纳入受试者 729 人,并已于今年 9 月完成试验。
疼痛是 DPNP 常见的临床症状,约见于 50% 左右的糖尿病和约 13% 的糖耐量受损的患者,严重影响其正常生理和精神状态,出现睡眠障碍、营养失调、运动受限、情感障碍,从而降低生活质量和工作能力。
DPNP 为排他性诊断,在糖尿病或糖尿病前期基础上,根据临床表现、神经系统查体及神经电生理检查证实存在周围神经病变。
目前尽管国内外指南推荐多种镇痛药物选择,DPNP 患者对镇痛治疗的反应程度不尽相同,无论对单药或联合治疗,部分患者疼痛控制均不理想,较大剂量用药也带来安全性问题。而对于国内患者而言,选择更少,同类药物普瑞巴林 DPNP 适应症及迈瑞巴林均未在国内批准上市。本次 HSK16149 若能顺利申报上市,无疑将为国内患者带来新的治疗选择。
除 DPNP 外,HSK16149 还在拓展多项其他适应症,针对带状疱疹后神经痛的 III 期临床研究也已于今年 7 月完成所有受试者的入组工作(登记号:
CTR20212551
)。而针对辅助镇痛、外周神经痛适应症目前也已进入临床阶段。
1、诺华:司库奇尤单抗新规格/新适应症在华获批上市
10 月 29 日,诺华中国宣布,其在研新药可善挺®(司库奇尤单抗)两种新规格获得国家药品监督管理局批准——300 mg 无忧随心笔®及 75 mg 预充注射针,儿童银屑病适应症扩展至符合系统治疗或光疗指征的中度至重度斑块状银屑病的 6 岁及以上患者,不再因体重受限。
银屑病是一种免疫相关的慢性、复发性、炎症性、系统性疾病。目前,我国有超过 650 万名银屑病患者。因疾病尚无法治愈,患者需长期遵循规范合理的治疗方案以获得良好的病情控制,达到皮损全清。在实际临床治疗中,坚持治疗对患者来说是一个极大的挑战。
2020 年 6 月,可善挺®150 mg 自感随心笔®在华获批,患者经过医师指导后在家即可自行注射,在使用过程中不会看到针头,降低注射焦虑,极大提升了患者的依从性和治疗体验。多数银屑病患者的常规治疗剂量为每次 300 mg,即单次 2 针 150 mg 皮下注射。
本次 300 mg 无忧随心笔®的获批,使银屑病患者通过更少的注射次数来实现满意的症状控制,即由单次 2 针 150 mg 简化为单次 1 针 300 mg。更简化的治疗方式给患者带来更轻松的治疗感受。另外,研究显示,300 mg 无忧随心笔®疗效和安全性与 2 针 150 mg 相当,注射部位不良反应发生率不到 0.1%。
此外,自 2021 年可善挺®获批儿童及青少年银屑病适应症以后,体重小于 50 kg 的患儿受限于体重而无法使用,很多家长在期盼着更小规格的产品能尽早获批上市。75 mg 新规格的获批给更多患儿家庭带来希望,可有效改善中重度银屑病患儿整体生活质量。
在安全性方面,研究显示,可善挺®治疗银屑病 12 周不良事件与安慰剂相似,持续接受治疗 52 周,无论是低剂量治疗组还是高剂量治疗组,未发现新增/非预期安全问题。
随着新规格的获批,可善挺®成为目前我国唯一获批三种规格(300 mg、150 mg、75 mg)的全人源白介素类抑制剂,患者可在医生的建议下根据自身需求选择不同规格的产品,单次治疗只需注射一针,有助于促进患者坚持规范用药进而实现更好的生活质量。
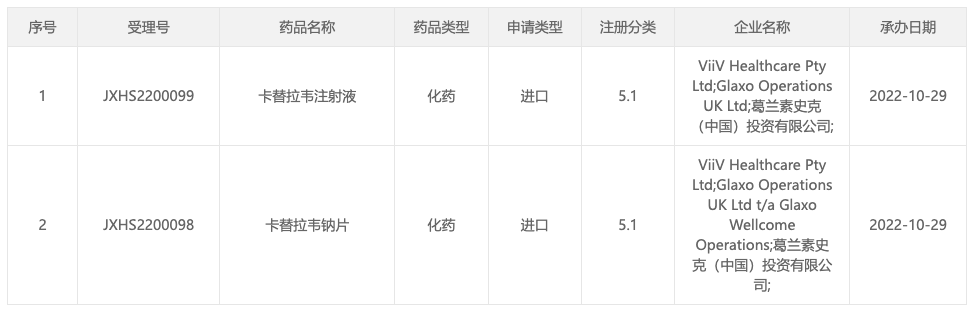
1、GSK/ViiV Healthcare:卡替拉韦国内申报上市
卡替拉韦(Cabotegravir)是一款长效 HIV-1 整合酶链转移抑制剂(INSTI),通过阻止病毒 DNA 整合到人类免疫细胞(T 细胞)的遗传物质中来抑制 HIV 复制。
据 Insight 数据库显示,卡替拉韦此前已于 2020 年 12 月获 EMA 批准上市,此后又先后于美国、日本获批上市。在国内,于 2021 年 11 月首次申报上市。
据此前披露的关键 III 期 ATLAS 和 FLAIR 研究结果显示,在为期 48 周的治疗期间,每月臀部肌肉注射一次持续病毒抑制效果与每日口服三联疗法(2 款核苷逆转录酶抑制剂(NRTIs)联用整合酶抑制剂 (INI),NNRTI,或蛋白酶抑制剂 (PI))相同。且有 90% 受试者表示与之前接受每日口服疗法相比更喜欢每月注射 1 次的疗法。
同样在本周,ViiV Healthcare 宣布 Cabotegravir 长效注射剂的上市许可申请已获 EMA 受理,用于暴露前预防性用药(PrEP),以降低性获得的 HIV-1 风险。在预防 HIV 方面,启动治疗后,Cabotegravir 长效注射剂每 2 个月注射一次,全年仅需注射 6 次。
此前 Cabotegravir 长效注射剂(CAB LA)用于 PrEP,已在美国、澳大利亚、津巴布韦获得批准,商品名为 Apretude。
本周境外共有 9 款新药(含改良新)研发进度推进到新阶段。
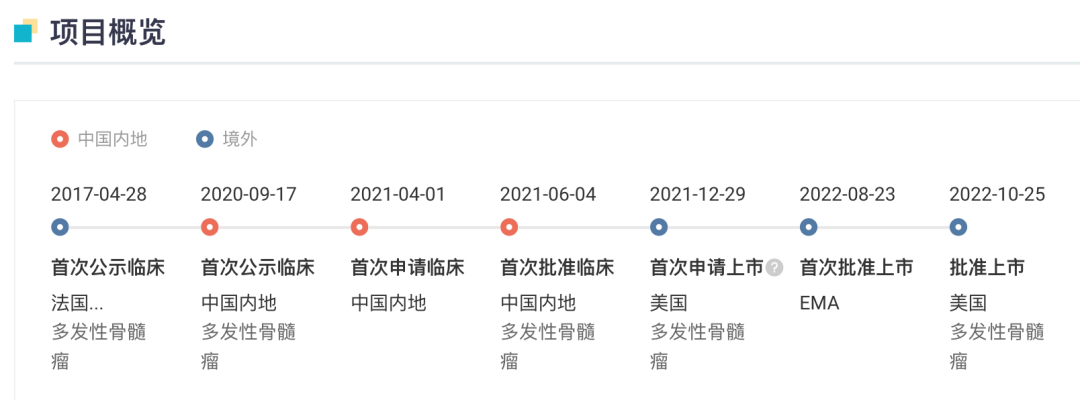
1、强生: BCMA × CD3 双抗获 FDA 批准上市
10 月 25 日,强生制药宣布其 BCMA × CD3 双抗 Teclistamab(商品名:Tecvayli)获 FDA 批准在美国上市,用于治疗既往接受过四线及以上治疗(包括蛋白酶体抑制剂、免疫调节剂和抗 CD38 单抗)的复发/难治性多发性骨髓瘤(R/R MM)。
 来自:Insight 数据库网页版
Teclistamab 的批准是基于 I/II 期临床试验 MajesTEC-1 研究(试验登记号:NCT04557098/NCT03145181)。这是一项单臂、开放标签、多队列、多中心剂量递增研究,以 ORR 为主要终点。
关键 II 期试验纳入既往接受过中位治疗线数五线治疗的患者(n=110),其中 78% 的患者都接受过四线及以上治疗。总缓解率(ORR)达到 61.8%(95% CI:52.1%,70.9%),其中 28.2% 的患者完全缓解(CR)或严格完全缓解(sCR)。
首次响应中位时间为 1.2 个月(范围:0.2 - 5.5 个月)
。中位随访 7.4 个月,预估的 6 个月持续缓解(DOR)率为 90.6%(95% CI:80.3%,95.7%),9 个月 DOR 率为 66.5%(95% CI:38.8%,83.9%)。
值得一提的是,这是强生获批的第 4 款多发性骨髓瘤疗法
。
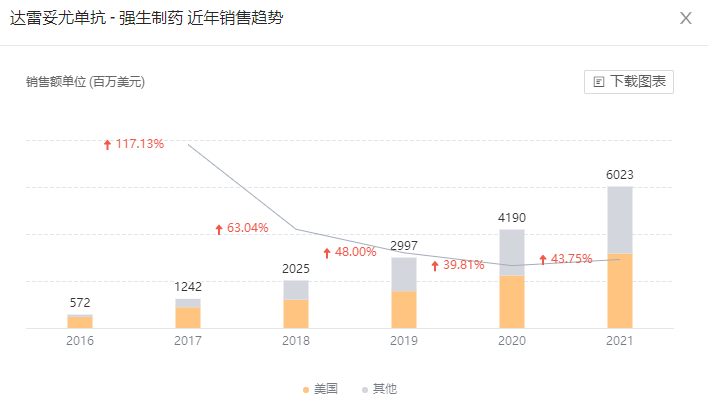
强生已经获批的四款多发性骨髓瘤药物中,CD38 单抗达雷妥尤单抗是 2021 年度全球销售 60.23 亿美元的超级重磅炸弹,且仍在以 43% 的高增速,势如破竹持续增长;CAR-T 疗法西达基奥仑赛尽管今年 2 月刚刚获批,但大有后来居上之势,Q3 单季度销售额就已经达到 5500 万美元,还在高速放量期。
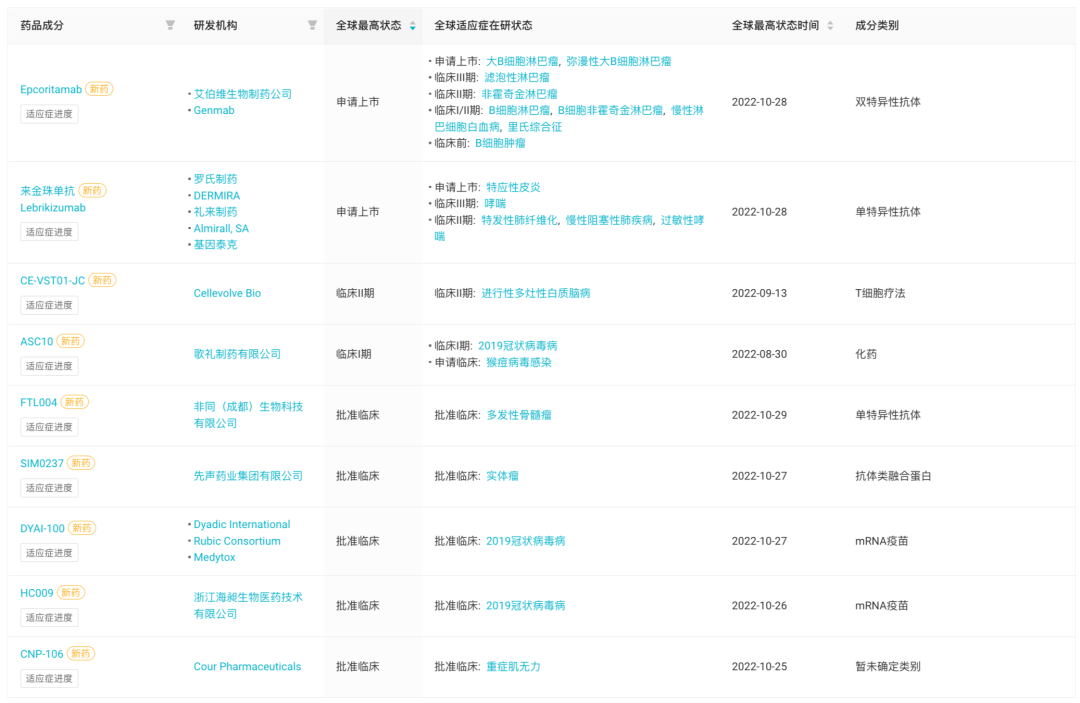
1、艾伯维/Genmab:CD3/CD20 双抗申报上市
10 月 28 日,艾伯维/Genmab 宣布已向 FDA 提交了 Epcoritamab 的生物制品许可申请(BLA),用于治疗此前接受 2 线或多线系统治疗后的复发/难治性大 B 细胞淋巴瘤(LBCL)成人患者。
此外,EMA 已受理 Epcoritamab 的营销授权申请(MAA),用于治疗接受 2 线或多线系统治疗的复发/难治性(R/R)弥漫性大 B 细胞淋巴瘤(DLBCL)成人患者。
Eporitamab 是基于 Genmab 公司专有的 DuoBody 技术开发的一款 CD3/CD20 双特异性抗体。Genmab 的 DuoBody-CD3 技术旨在选择性地引导细胞毒性 T 细胞进入肿瘤,以引发对恶性细胞的免疫反应。Epcoritamab 被设计成可同时结合 T 细胞上的 CD3 和 B 细胞上的 CD20,并诱导 T 细胞介导的靶向淋巴瘤 B 细胞的杀伤作用。
Epcoritamab 此项监管申请是基于此前披露的一项 EPCORE NHL-1 II 期临床试验大细胞淋巴瘤扩展队列的初步结果。
该研究队列包括 157 例复发/难治性 LBCL 患者,这些患者此前接受过中位 3.5 线(2-11 线)治疗,其中 38.9% 的患者此前接受过 CAR-T 细胞治疗。 该队列的关键结果显示,独立审查委员会(IRC)证实的客观缓解率(ORR)为 63.1%,观察到的中位缓解持续时间(DoR)为 12 个月。
来自:Insight 数据库网页版
Teclistamab 的批准是基于 I/II 期临床试验 MajesTEC-1 研究(试验登记号:NCT04557098/NCT03145181)。这是一项单臂、开放标签、多队列、多中心剂量递增研究,以 ORR 为主要终点。
关键 II 期试验纳入既往接受过中位治疗线数五线治疗的患者(n=110),其中 78% 的患者都接受过四线及以上治疗。总缓解率(ORR)达到 61.8%(95% CI:52.1%,70.9%),其中 28.2% 的患者完全缓解(CR)或严格完全缓解(sCR)。
首次响应中位时间为 1.2 个月(范围:0.2 - 5.5 个月)
。中位随访 7.4 个月,预估的 6 个月持续缓解(DOR)率为 90.6%(95% CI:80.3%,95.7%),9 个月 DOR 率为 66.5%(95% CI:38.8%,83.9%)。
值得一提的是,这是强生获批的第 4 款多发性骨髓瘤疗法
。
强生已经获批的四款多发性骨髓瘤药物中,CD38 单抗达雷妥尤单抗是 2021 年度全球销售 60.23 亿美元的超级重磅炸弹,且仍在以 43% 的高增速,势如破竹持续增长;CAR-T 疗法西达基奥仑赛尽管今年 2 月刚刚获批,但大有后来居上之势,Q3 单季度销售额就已经达到 5500 万美元,还在高速放量期。
1、艾伯维/Genmab:CD3/CD20 双抗申报上市
10 月 28 日,艾伯维/Genmab 宣布已向 FDA 提交了 Epcoritamab 的生物制品许可申请(BLA),用于治疗此前接受 2 线或多线系统治疗后的复发/难治性大 B 细胞淋巴瘤(LBCL)成人患者。
此外,EMA 已受理 Epcoritamab 的营销授权申请(MAA),用于治疗接受 2 线或多线系统治疗的复发/难治性(R/R)弥漫性大 B 细胞淋巴瘤(DLBCL)成人患者。
Eporitamab 是基于 Genmab 公司专有的 DuoBody 技术开发的一款 CD3/CD20 双特异性抗体。Genmab 的 DuoBody-CD3 技术旨在选择性地引导细胞毒性 T 细胞进入肿瘤,以引发对恶性细胞的免疫反应。Epcoritamab 被设计成可同时结合 T 细胞上的 CD3 和 B 细胞上的 CD20,并诱导 T 细胞介导的靶向淋巴瘤 B 细胞的杀伤作用。
Epcoritamab 此项监管申请是基于此前披露的一项 EPCORE NHL-1 II 期临床试验大细胞淋巴瘤扩展队列的初步结果。
该研究队列包括 157 例复发/难治性 LBCL 患者,这些患者此前接受过中位 3.5 线(2-11 线)治疗,其中 38.9% 的患者此前接受过 CAR-T 细胞治疗。 该队列的关键结果显示,独立审查委员会(IRC)证实的客观缓解率(ORR)为 63.1%,观察到的中位缓解持续时间(DoR)为 12 个月。
1、先声药业: PD-L1/IL-15 双抗 IND 申请获 FDA 批准
10 月 27 日,先声药业宣布其自主研发的抗 PD-L1/IL-15 双特异性抗体 SIM0237 注射液新药临床试验申请 (IND) 获美国食 品药品监督管理局 (FDA) 批准,拟用于治疗局部晚期不可切除或转移性实体瘤。
SIM0237 是基于先声药业自有蛋白质工程技术平台开发的一种抗 PD-L1 单抗与 IL-15/IL-15Rα 融合蛋白,可通过结合 PD-L1 阻断 PD1/PD-L1 免疫抑制通路,同时通过 IL-15 激活免疫系 统,从而起到了解除免疫抑制和激活免疫系统的双重协同作用,发挥抗肿瘤作用。
临床前研究显示,SIM0237 在小鼠肿瘤模型中药效优于 PD-L1 单药和 IL-15 单药,有较高的临床开发潜力。
此前,SIM0237 在国内的临床试验申请已于今年 10 月 10 日获 CDE 受理(受理号:
CXSL2200508
)。
此外,上周美国 FDA 的心血管及肾病药物专家咨询委员会(CRDAC)召开了会议,讨论来自 GSK 的第 3 款口服小分子缺氧诱导因子脯氨酰羟化酶 (HIF-PH) 抑制剂「达普司他」的获益风险情况,以决定其治疗慢性肾病(CKD)相关贫血的 NDA 申请(216951)能否获得批准。
对于透析依赖患者,FDA 认为其疗效与成熟药物促红细胞生成素(ESA)相似,且未增加安全性风险,但首款口服药带来的意义存疑;对于非透析患者,FDA 表示达普司他增加了心血管、中风等额外风险,且口服药不利于疾病管理。
投票结果显示,
专家们对这两项适应症给予了相反的意见,其中,针对透析依赖患者群体的适应症赢得了专家们 13:3 的支持票,专家认为首款口服药对这部分未满足医疗需求的积极意义,在优异疗效之下,口服给药的便利性有助于减轻了医患负担和医疗成本。
而针对非透析依赖患者群体的适应症则被 11:5 致以反对,专家认为在安全性风险增加的情况下,获益不足以抵消。
从投票结果来看虽然喜忧参半,但若达普司他能以 DD-CKD 相关贫血适应症率先出道,将是 HIF-PHI 在美国市场的一步突破。
事实上以罗沙司他为例,该药在中国、日本也都是先在透析依赖患者中获得批准,再将适应症拓展至非透析依赖人群。
免责声明:
本文仅作消息分享,
并不构成投资建议,也不代表 Insight 数据库的立场,文章观点仅供分享行业见解,请广大投资者谨慎。
投稿:微信 insightxb;邮箱 insight@dxy.cn
免费试用 Insight 数据库



 个人中心
个人中心
 我是园区
我是园区
 退出
退出