

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

异位骨化(HO)是指在非骨骼组织(包括肌肉、肌腱或其他软组织)中出现病理性骨组织形成【1】。根据成因可分为获得性及遗传性两大类型,遗传性异位骨化主要包括进行性肌肉骨化症 (fibrodysplasia ossificans progressive, FOP)【2】和进行性骨异位生长 (progressive osseous heteroplasia, POH)【3】两种类型。获得性异位骨化主要继发于创伤、烧伤、神经损伤,手术以及关节置换术后,是临床上严重的并发症,其发病机制尚未明了。目前,由于治疗方法有限,且手术治疗受到高复发率和并发症的限制,治疗疗效往往不尽人意。过去的研究都集中于某一种异位骨化,缺乏对异位骨化机制的深入统一认识。
2021年6月23日,美国哈佛大学杨英姿教授领导的科研团队在Science Translational Medicine杂志上发表了题为 A self-amplifying loop of YAP and SHH drives formation and expansion of heterotopic ossification的研究论文(论文第一作者为丛茜博士),揭示了获得性及遗传性异位骨化形成及扩展的共同分子机制,为预防及治疗异位骨化提供了重要的细胞和分子基础。

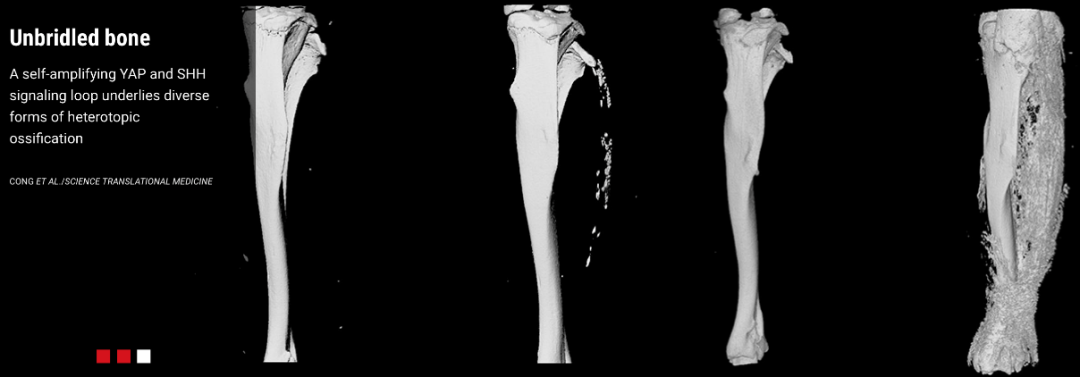
进行性骨异位生长 (POH) 是由GNAS基因失活突变所引起的异位骨化。临床上,POH 的患者在婴儿期皮肤开始骨化,儿童期皮下和深层结缔组织异位骨化呈镶嵌分布并逐步扩大,导致关节强直及肢体生长迟缓【4】。在POH的小鼠模型中杨教授团队发现异位骨化的不断扩大是由于Gnas缺失的细胞通过分泌SHH不断招募周围的正常细胞, 并且诱导这些细胞分化为成骨细胞,促进了异位骨的不断扩大(如图1所示)。这一细胞非自主性的调控过程揭示了异位骨化不断增大扩展的重要细胞机制。
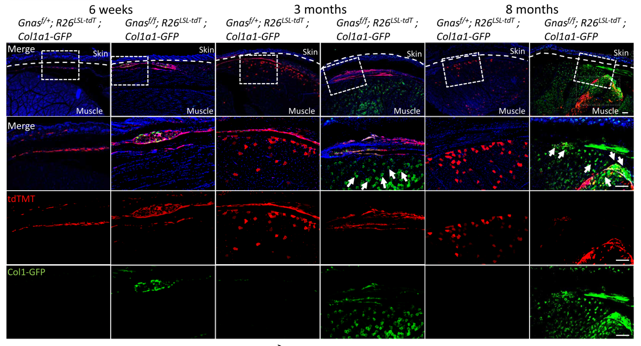
Figure 1. Col-GFP 及 tdTMT 在POH小鼠模型中的免疫荧光染色图。
随后,杨教授团队通过RNA-seq分析发现Gnas的失活导致YAP转录活性的增加,这表明了YAP很可能是Shh基因表达及分泌增加的重要调控因子。并且一系列的细胞及分子实验证实了YAP与SHH可相互调控,形成正反馈调控回路。为了进一步研究异位骨化发生的分子机制,该团队对Gnas缺失的细胞及对照组细胞用染色质转座酶可及性测序(ATAC-seq)分析进行全基因组染色质重塑研究,发现在Gnas缺失的细胞中基因的转录活性增强,并且在Shh启动子及增强子区域DNA的开放增强。进一步的分析找了YAP/TEAD4的可能结合位点并由ChIP-PCR证实(如图2所示)。为了检验已找到的YAP/TEAD4结合位点是否是Shh表达所必需的,杨教授团队通过CRISPRi技术【5,6】靶向抑制了YAP/TEAD4与已知的Shh启动子及增强子位点的结合,发现Shh表达水平显著下降,从而证实了YAP直接转录调控Shh的表达(如图2所示)。
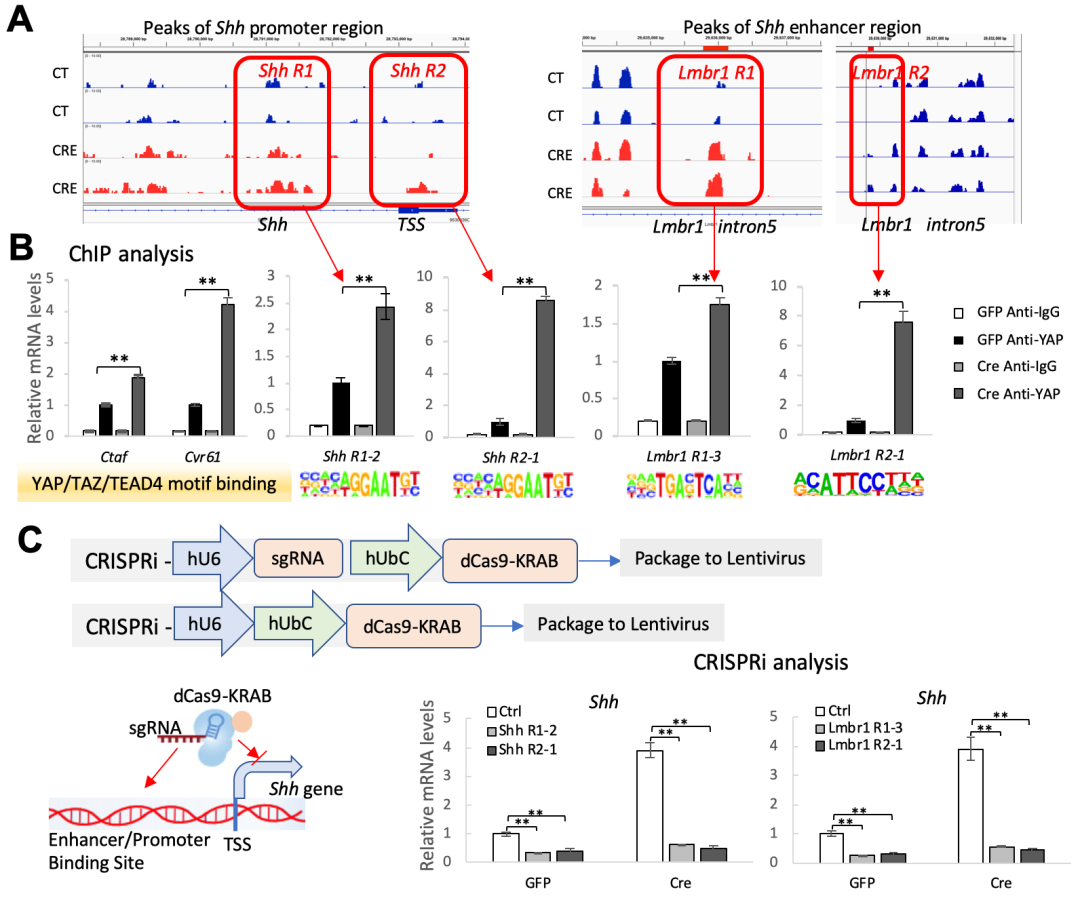
Figure 2. (a) ATAC-seq,(b) ChIP-PCR 及(c) CRISPRi分析结果。
进行性骨化性纤维发育不良(FOP)是另一种罕见的遗传性异位骨化,是由I 型BMP 受体(ACVR1)的激活突变引起的【7】。临床上,FOP的异位骨化首先发生在躯体的背侧、中轴、头部及肢体近端部位,逐渐发展到躯体的腹侧、附件、尾侧和肢体远端。为了验证是否YAP-SHH正反馈调控回路同样在FOP模型中起到关键作用,该团队分别在FOP小鼠模型中敲了Yap及Shh,发现Yap或Shh敲除后异位骨化明显减少甚至完全消失(如图3所示),从而证实了YAP-SHH正反馈调控回路在FOP中的关键作用。
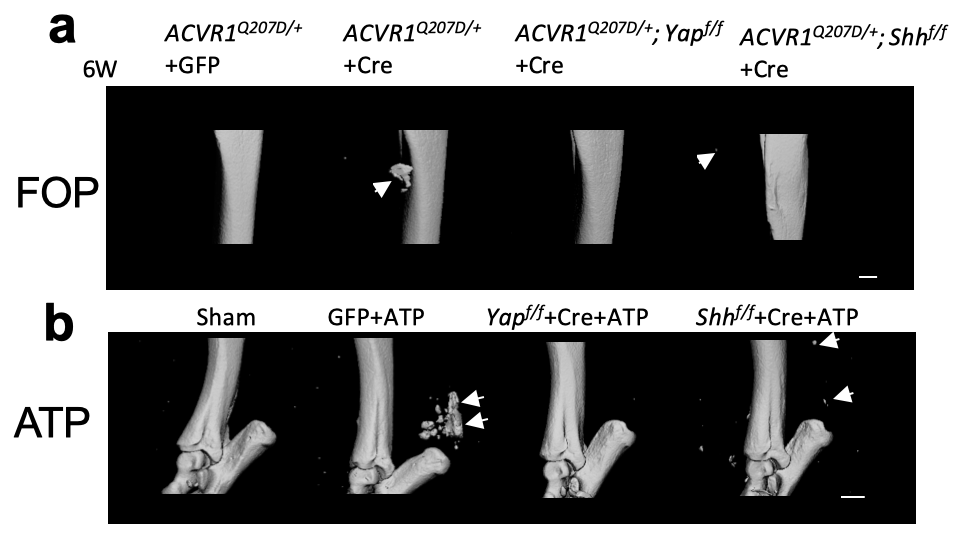
Figure 3. (a) FOP及(b) ATP小鼠模型中敲除Yap及Shh异位骨化减少。
获得性异位骨化是临床上常见的并发症,其发病率高治疗难度大。通过对遗传性异位骨化的研究为更常见的获得性异位骨化的发病机制及治疗提供了很好的基础。杨教授团队在小鼠中成功建立了损伤诱导的异位骨化模型, 跟腱穿刺骨化(ATP)模型【8】,并发现YAP及SHH在跟腱损伤后表达上调。在跟腱中敲除Yap或Shh可以显著减少或完全阻止异位骨化的产生(如图3所示)。在跟腱损伤后使用YAP抑制剂或SHH单克隆抗体治疗可以有效减少甚至抑制异位骨的产生。
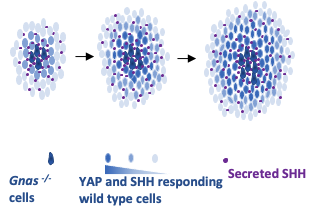
Figure 4. YAP-SHH自放大正反馈回路调控异位骨化发生及发展。
综上所述,杨教授团队揭示了YAP-SHH细胞自放大及自传播的正反馈回路是不同种类异位骨化形成及扩展的共同的核心机制(如图4所示)。该YAP-SHH正反馈回路不仅在两种遗传性异位骨化中调控异位骨的产生及扩展,而且在损伤诱导的异位骨化中同样至关重要。在异位骨化病人样本中同样发现了YAP及SHH的表达上调。因此,该团队的研究结果为有效治疗人类罕见病中和常见的损伤引起的异位骨化带来了希望。
原文链接:
https://stm.sciencemag.org/content/13/599/eabb2233
参考文献
1. Rigaux, P., et al., Study of serum factors potentially involved in the pathogenesis of heterotopic bone formation after severe brain injury. Joint Bone Spine, 2005. 72(2): p. 146-9.
2. Shafritz, A.B., et al., Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Engl J Med, 1996. 335(8): p. 555-61.
3. Kaplan, F.S. and E.M. Shore, Progressive osseous heteroplasia. J Bone Miner Res, 2000. 15(11): p. 2084-94.
4. Shore, E.M., et al., Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med, 2002. 346(2): p. 99-106.
5. Yeo, N.C., et al., An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods, 2018. 15(8): p. 611-616.
6. Thakore, P.I., et al., Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods, 2015. 12(12): p. 1143-9.
7. Hatsell, S.J., et al., ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med, 2015. 7(303): p. 303ra137.
8. Wang, X., et al., Inhibition of overactive TGF-beta attenuates progression of heterotopic ossification in mice. Nat Commun, 2018. 9(1): p. 551.
转载须知
【非原创文章】本文著作权归文章作者所有,欢迎个人转发分享,未经允许禁止转载,作者拥有所有法定权利,违者必究。







