

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

01
Evinacumab
2021年2月11日,FDA首次批准了Evinacumab-dgnb(商品名EVKEEZATM),用于12岁及以上儿童或成人家族性纯合子高胆固醇血症(HoFH)患者治疗。
Evinacumab是一种全人源靶向血管生成素样蛋白3(ANGPTL3)的IgG4亚型的单克隆抗体,能结合并抑制ANGPTL3。ANGPTL3是血管生成素样蛋白家族的成员,主要在肝脏表达,通过抑制脂蛋白脂酶(LPL)和内皮脂酶(EL)在调节脂质代谢中发挥作用。evinacumab抑制ANGPTL3通过挽救LPL和EL的活性降低LDL-C、HDL-C和甘油三酯(TG)。
此前,FDA授予了evinacumab突破性药物资格(BTD),孤儿药资格和优先审评资格,此次批准是基于III期ELIPSE-HoFH研究(NCT03399786)的积极结果。evinacumab的推荐剂量为15 mg/kg静脉输注,每4周1次,由Regeneron开发。
Evinacumab常见不良反应(≥5%)为鼻咽炎、流感样疾病、头晕、鼻漏和恶心。
02
Dostarlimab
2021年4月22日, FDA首次批准了dostarlimab-gxly(商品名JEMPERLI),作为一种单药疗法,用于治疗接受含铂化疗期间或之后病情进展、错配修复缺陷(dMMR)复发性或晚期子宫内膜癌患者。
Dostarlimab是一款人源化抗PD-1单克隆抗体,与PD-1受体结合,并阻断其与配体PD-L1和PD-L2的相互作用,值得一提的是,dostarlimab是第一个被批准用于治疗子宫内膜癌的PD-1疗法,同时也是FDA批准的第七款PD-(L)1药物。
此前,FDA授予了dostarlimab突破性药物资格(BTD)和优先审评资格,此次批准是基于单组、多队列GARNET研究数据(NCT02715284)的积极结果。
Dostarlimab最常见的不良反应(≥20%)为疲劳/乏力、恶心、腹泻、贫血和便秘。此外,Dostarlimab可引起免疫介导性副作用的严重疾病,包括健康器官的炎症,如肺炎、结肠炎、肝炎、内分泌疾病和肾炎。
03
Loncastuximab tesirine
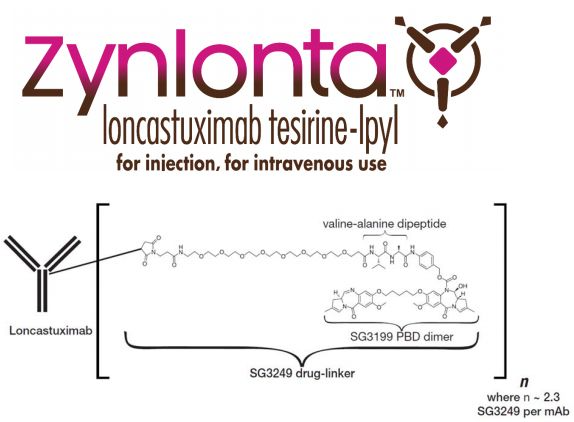
2021年4月23日, FDA首次批准了loncastuximab tesirine-lpyl(商品名Zynlonta),用于治疗已接受过2种或多种系统疗法的复发或难治性(r/r)大B细胞淋巴瘤(LBCL)成人患者,包括弥漫性大B细胞淋巴瘤(DLBCL)、起源于低级别淋巴瘤和高级别细胞淋巴瘤的DLBCL。
Zynlonta是一种靶向CD19的抗体偶联药物(ADC),由一种人源化抗人CD19单克隆抗体通过连接器与吡咯并苯并二氮杂卓(pyrrolobenzodiazepine,PBD)二聚体细胞毒素偶联而成。一旦与表达CD19的细胞结合,loncastuximabtesirine就会被细胞内化,随后释放出细胞毒素,该毒素能不可逆地与DNA结合,从而产生阻止DNA链分离的强力链间交联,从而破坏复制等必要的DNA代谢过程,最终导致细胞死亡。
此前,Zynlonta通过优先审查程序获得加速批准,此次加速批准是基于关键LOTIS-2临床试验的积极数据。
在汇总的安全性人群中,Zynlonta最常见的不良反应(≥20%)为血小板减少、γ-谷氨酰转移酶升高、中性粒细胞减少、贫血、高血糖、转氨酶升高、疲劳、低蛋白血症、皮疹、水肿、恶心和肌肉骨骼疼痛。
04
Amivantamab

2021年5月21日, FDA首次批准了amivantamab-vmjw(商品名RYBREVANT),用于治疗在接受含铂化疗期间或之后病情进展、表皮生长因子受体(EGFR)基因第20号外显子有插入突变的转移性非小细胞肺癌(NSCLC)患者。
Amivantamab是一种同时靶向EGFR和cMET的双特异性抗体,它能够通过阻断配体结合和在外显子20插入突变模型中降解EGFR和MET来破坏EGFR和MET信号功能,EGFR和MET在肿瘤细胞表面的存在也使得免疫效应细胞(如自然杀伤细胞和巨噬细胞)分别通过抗体依赖性细胞毒性(ADCC)和吞噬机制靶向肿瘤细胞进行杀伤。
值得一提的是,amivantamab是FDA批准的用于治疗EGFR外显子20插入突变的首个靶向治疗药物。此次批准,是基于I期CHRYSALIS研究(NCT02609776)的积极结果。amivantamab的推荐剂量基于基线体重,前4周每周一次给药,第一次给药分2天给完,4周后维持每2周一次给药。
在接受RP2D剂量amivantamab治疗的患者(n=114)中,最常见的治疗期间出现的不良事件(TEAE)是皮疹(86%)、输液相关反应(IRR,66%)、甲沟炎(45%)。其他不良事件为口腔炎(21%)和瘙痒(17%)。
05
Aducanumab

2021年6月7日, FDA首次批准了aducanumab-avwa(商品名ADUHELM),用于治疗阿尔茨海默症患者。
Aducanumab是一种靶向β淀粉样蛋白的单克隆抗体,β淀粉样蛋白斑块在大脑种的积累是阿尔茨海默症的一个重要病理生理特征。aducanumab通过加速审批程序获得批准,是自2003年以来美国FDA批准的首个治疗AD的新药。此次批准是基于3项独立的临床试验结果,达到了大脑中淀粉样β斑块减少的替代终点,这些结果支持了Aduhelm的加速批准。aducanumab的推荐剂量为10mg/kg,每4周一次给药。
此前,FDA已授予aducanumab快速通道资格(FTD)。根据加速批准条款,FDA已要求渤健开展一项新的随机对照临床试验,以验证该药物的临床疗效。如果试验未能证实临床疗效,FDA可能会启动程序撤销对该药物的批准。
Aducanumab最常见的不良反应(与安慰剂相比至少10%或更高的发生率):ARIA水肿、头痛、ARIA-H微出血、ARIA-H浅表铁质沉着和跌倒。
06
Anifrolumab

2021年7月30日, FDA首次批准了anifrolumab-fnia(商品名SAPHNELO),用于正在接受标准疗法的中度至重度系统性红斑狼疮(SLE)成人患者的治疗。
Anifrolumab是一种靶向 I型IFN受体的亚基1的全人源单克隆抗体,可阻断I型IFN的活性。I型IFN如IFN-α、IFN-β和IFN-κ是参与调节SLE炎症通路的细胞因子。I型IFN在SLE的发病机制中起着重要作用,大约60-80%的活动性SLE成年患者表达I型IFN诱导基因水平升高。
此次批准,基于两项名为TULIP的3期临床试验(NCT02446912和NCT02446899)以及名为MUSE的2期临床试验(NCT01438489)的疗效和安全性数据。Anifrolumab推荐剂量为每4周300mg静脉滴注。
Anifrolumab由阿斯利康于2004年通过与Medarex公司的独占许可(exclusive license)和合作协议获得了全球权利。anifrolumab用于治疗SLE的许可也正在欧盟和日本接受监管审评,在国内,阿斯利康按治疗用生物制品1类递交的Saphnelo注射液临床试验申请获得默示许可,拟开发用于中度至重度活动性系统性红斑狼疮的治疗。
Anifrolumab最常见的药物不良反应(发生率≥5%)为鼻咽炎,上呼吸道感染、支气管炎、输液相关反应、带状疱疹和咳嗽。
07
Tisotumab vedotin
2021年9月20日, FDA首次批准了Tisotumab vedotin-tftv(商品名TIVDAK),用于在化疗期间或化疗后病情进展的复发性或转移性宫颈癌患者。
Tisotumab vedotin是一种靶向组织因子(tissuefactor,TF)的抗体药物偶联物(ADC),TF是一种参与肿瘤信号传导和血管生成的蛋白质,在绝大多数宫颈癌患者和许多其他实体瘤(包括卵巢、肺、胰腺、结直肠和头颈部癌症)中过度表达。基于TF因子在许多实体瘤中的高表达和快速内化,使TF成为了开发ADC药物的理想靶标。
此次批准,基于关键II期innovaTV204研究的疗效和安全性数据。Tisotumabvedotin推荐剂量为每3周2mg/kg静脉滴注。
Tisotumab vedotin由由西雅图遗传学公司和Genmab公司联合开发,结合了Genmab公司的TF靶向单抗Tisotumab以及西雅图遗传学公司的ADC技术。
Tisotumab vedotin最常见的(≥25%)不良反应,包括血红蛋白减少、疲劳、淋巴细胞减少、恶心、周围神经病变、脱发、鼻出血、结膜不良反应、出血、白细胞减少、肌酐增加、干眼症、凝血酶原国际标准化比率增加、活化部分凝血活酶时间延长,腹泻和皮疹。
08
Tezepelumab
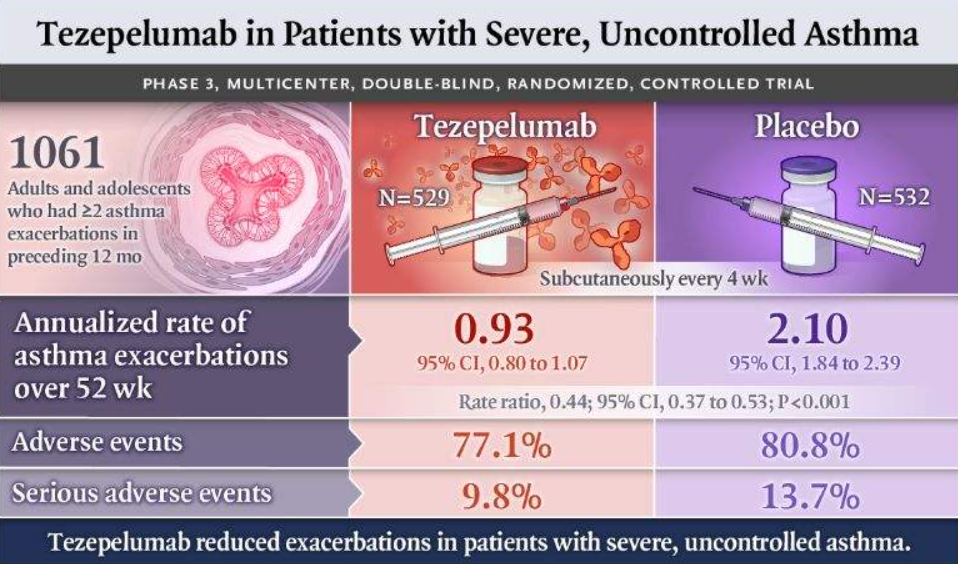
2021年12月17日, FDA首次批准了Tezepelumab-ekko(商品名Tezspire),作为一种附加维持疗法,用于治疗年龄≥12岁的严重哮喘儿科患者和成人患者。
Tezepelumab是一种靶向胸腺间质淋巴细胞生成素(TSLP)的单克隆抗体,TSLP是一种上皮细胞因子,在哮喘炎症中起关键作用。Tezspire通过优先审查程序获得批准。在2018年9月,美国FDA授予了tezepelumab治疗无嗜酸性粒细胞表型严重哮喘的突破性药物资格(BTD)。值得一提的是,在治疗严重哮喘方面,Tezspire是唯一一个没有表型(如嗜酸性粒细胞或过敏)或生物标志物限制的生物制剂。
此次批准,基于关键性III期NAVIGATOR试验的疗效和安全性数据。Tezepelumab推荐剂量为每4周210mg皮下注射。Tezepelumab由安进和阿斯利康公司联合开发。
Tezepelumab最常见的不良反应(≥ 3%)是咽炎、关节痛和背痛。
声明:本稿件为转载,仅用于分享,不代表本公众号立场,如涉及版权等问题,请尽快联系我们,我们将第一时间更正或删除,谢谢
往期回顾
|
1 |
|
|
2 |
|
|
3 |
|

媒体合作:李先生 13173667890
商务合作:李小姐 L13695604846(微信)
● 扫码关注我们
· BioClub ·






