

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

2021年10月22日,来自中国科学院上海药物研究所的蒋华良院士和郑明月研究员课题组在Protein & Cell上发表了文章 Drug target inference by mining transcriptional data using a novel graph convolutional network framework 。研究人员应用孪生谱图卷积网络(SSGCN),开发了一种新颖的基于转录组数据的药物靶标预测算法,并对模型的预测结果进行了湿实验验证。结果表明,SSGCN模型可以显著地提升药物靶标预测精度,为药物作用机制研究和靶标确证提供了有力手段【1】。

目前大多数药物都是通过与其体内的特定靶标相互作用来发挥其治疗作用。明确药物作用靶点和作用机制,对于药物研究开发和上市申请至关重要。对于表型筛选得到的活性化合物或天然产物活性成分,作用靶标的发现确证是进一步研究和开发的关键难点。除了加快早期的药物发现过程,对药物潜在靶标的识别还能加深我们对药物的作用机制、代谢、不良反应和耐药性的理解,为发现药物的新适应症指明方向,显著降低药物研发的周期和成本【1】。
药物靶标可以通过生化实验(例如蛋白质组学质谱法)进行识别,然而,受实验规模、精度和费用等因素的制约,大规模的实验通常难以广泛应用。而基于计算的靶标预测方法,有其低成本和高通量的特性,因此一直备受关注。这些方法对于靶向性药物开发、天然产物活性成分的作用机理,以及化学生物学研究都具有十分重要的意义。经典的药物靶标预测算法一般包括基于配体的方法和基于受体蛋白质结构的方法: 前者主要利用小分子结构或者理化性质,如分子指纹、形状和药效团来预测药物-靶点相互作用,而后者通常依靠分子对接来揭示小分子和蛋白质之间的潜在的相互作用。在前期工作中,上海药物所蒋华良院士和华东理工大学李洪林团队在基于配体和蛋白质受体结构的靶标预测方面开展了深入的研究【3-5】。
近年来,多组学数据的快速积累以及人工智能技术的发展,为开发更精准的药物靶标推理算法奠定了基础。其中,药物相关的基因表达谱数据,可以看做是从基因和细胞层面刻画药物的作用特征,因此对于药物靶标预测具有重要的参考价值。例如,Broad institute开发的Connectivity Map(CMap)方法是基于差异表达的特征基因间的相似性分析,为药物重定向、药物靶标和作用机制推理提供了重要线索【6】。此外还有一些基于动态网络分析和机器学习的方法,但生物网络的浅层分析方法一般难以发现化合物、基因扰动后转录谱之间的深度相关性。总体而言,药物作用在细胞水平引起的特征基因变化为靶标预测提供了丰富的信息,但如何透过高维度、高冗余度、以及高噪声的基因表达谱数据发现该药物的真实物理作用靶点仍然是一个尚未解决的重要挑战。比如,如何系统地考虑生物调控网络中基因的关联关系,如何考虑由细胞内噪声、细胞间差异、化合物作用浓度、作用时间等复杂因素对表达谱的影响,以及如何扩展可推理的药物靶点范围,等等。
为了应对上述挑战,科研人员基于对比学习和度量学习的思想设计了孪生网络架构,基于转录组数据预测靶标。该模型利用两个平行的图卷积网络分别从化合物微扰和基因微扰诱导的差异基因表达谱中提取特征,有效的降低基因表达谱中噪声对药物-靶标作用预测的影响。与已有的靶标预测算法比较,在基准数据集上,该方法的Top 100准确度达到了0.53,显著高于CMap方法【3】。通过利用深度学习挖掘转录组数据和蛋白调控网络,SSGCN方法引入的假设更少,同时可以学习化合物微扰谱和基因微扰谱的深层相关性。研究团队分析发现,传统的生物信息学方法(例如皮尔逊相关系数和KEGG特征谷本相关系数)难以捕捉这种深层相关性,这也解释了模型为什么取得了显著的性能提升。此外,该方法还整合了异质实验条件信息(细胞类型,持续时间和化合物剂量),不但可以利用更多的训练数据来提高模型表现,还可以考虑细胞系背景,剂量和时间依赖性等效应对差异基因表达,更好地考虑复杂的网络扰动对药物靶标预测推理的影响。
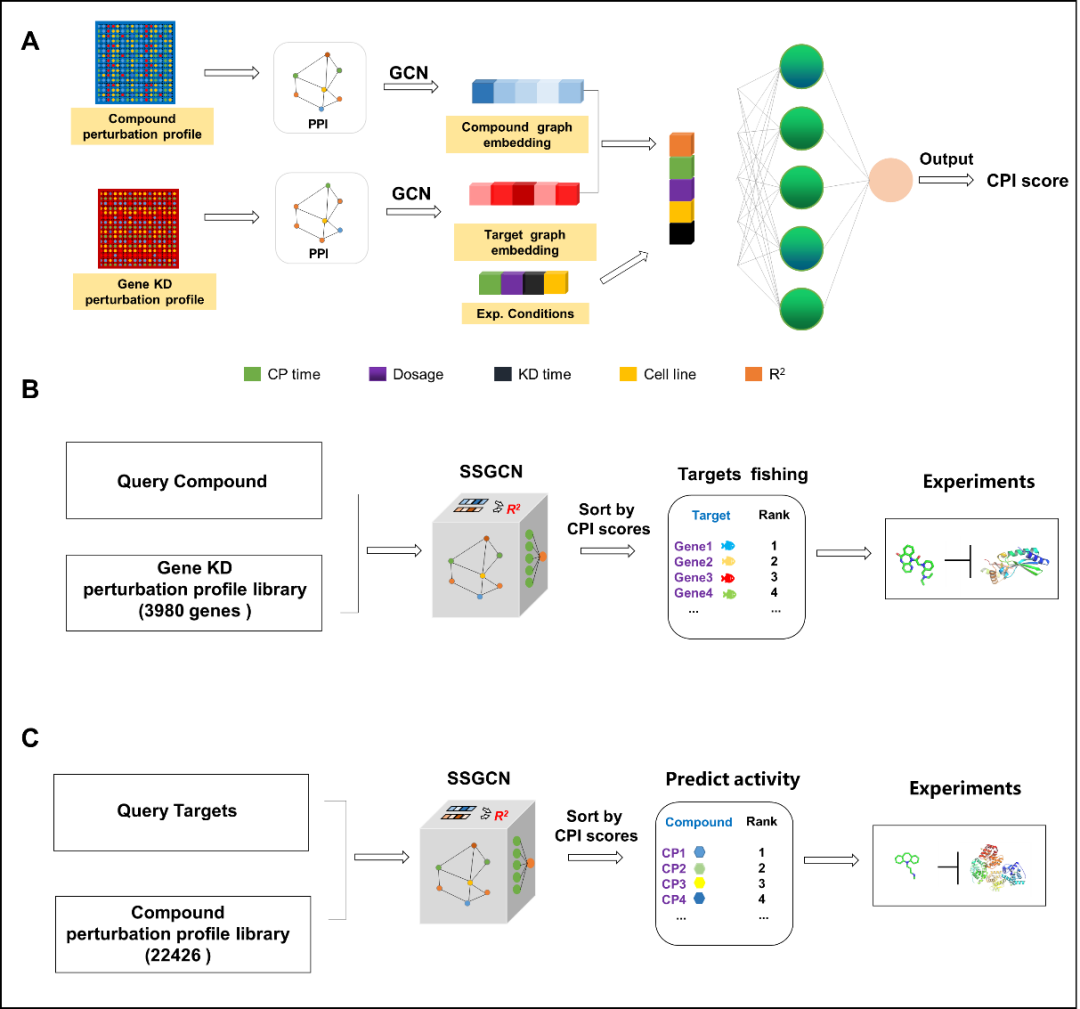 图1.靶标预测使用SSGCN模型
图1.靶标预测使用SSGCN模型
此外,为了进一步验证该方法,研究团队还利用该方法开展了湿实验的应用研究。在第一种应用场景中,研究人员建立了以化合物为中心的靶标推理流程预测奈非那韦(Nelfinavir, NFV)的潜在宿主靶标。实验结果成功验证了亲环蛋白A (cyclophilin A, CYPA) 是 NFV 的作用靶点,解释了NFV抗新冠病毒活性【7】的可能作用机制;在第二种应用场景中,研究团队建立了以靶标为中心的预测流程筛选外核苷酸焦磷酸酶/磷酸二酯酶1 (Ectonucleotide pyrophosphatase/phosphodiesterase 1, ENPP1) 的抑制剂。实验结果成功发现并确证了老药甲氨蝶呤 (Methotrexate, MTX)是一种新骨架的ENPP1抑制剂。
目前,人工智能已经在蛋白质结构预测、药物分子生成和反应路线规划等领域获得了巨大的成功。本项工作基于转录组数据预测药物靶标,在细胞环境中对药物作用模式进行了表征,从细胞转录组学和RNA生物学的角度探索药物作用的潜在靶标,是人工智能助力药物研发的一次积极尝试。
中国科学院上海药物研究所药物设计与发现中心的郑明月研究员、张素林副研究员和蒋华良院士为论文共同通讯作者,博士研究生钟飞盛、吴小龙和杨瑞瑞为论文共同第一作者。
原文链接:
https://doi.org/10.1007/s13238-021-00885-0
参考文献
-
Zhong FS, Wu XL, Yang RR, et al. Drug target inference by mining transcriptional data using a novel graph convolutional network framework [J]. Protein & Cell, 2021, (in press) https://link.springer.com/article/10.1007/s13238-021-00885-0 -
Keiser M J, Setola V, Irwin J J, et al. Predicting new molecular targets for known drugs [J]. Nature, 2009, 462(7270): 175-181. -
Li H, Gao Z, Kang L, et al. TarFisDock: a web server for identifying drug targets with docking approach [J]. Nucleic acids research, 2006, 34(suppl_2): W219-W224. -
Liu X, Gao Y, Peng J, et al. TarPred: a web application for predicting therapeutic and side effect targets of chemical compounds [J]. Bioinformatics, 2015, 31(12): 2049-2051. -
Wang X, Shen Y, Wang S, et al. PharmMapper 2017 update: a web server for potential drug target identification with a comprehensive target pharmacophore database [J]. Nucleic acids research, 2017, 45(W1): W356-W360. -
Subramanian, A., Narayan, R., Corsello, S.M., Peck, D.D., Natoli, T.E., Lu, X., Gould, J., Davis, J.F., Tubelli, A.A. and Asiedu, J.K. (2017) A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell, 171, 1437-1452. -
Xu Z, Yao H, Shen J, et al. Nelfinavir is active against SARS-CoV-2 in Vero E6 cells. (2020) Preprint at https://chemrxiv.org/articles/Nelfinavir_Is_Active_Against_SARS-CoV-2_in_Vero_E6_Cells/12039888 [J].
转载须知
【非原创文章】本文著作权归文章作者所有,欢迎个人转发分享,未经允许禁止转载,作者拥有所有法定权利,违者必究。







