Nature Immunol | 吴博文等揭示天冬氨酸代谢异常参与自身免疫疾病的发生
收藏
关键词:
Nature免疫揭示Nat疾病
资讯来源:BioArt + 订阅账号
发布时间:
2021-11-26

责编 | 兮
类风湿性关节炎
(
RA
)
是常见的自身免疫性关节炎,其特征是早期对瓜氨酸蛋白的免疫耐受丧失,最终导致关节炎症和破坏
【1】
。肿瘤坏死因子
(TNF)
是 RA 的关键炎性细胞因子,滑膜内 T 细胞是主要的 TNF 生产者
【2】
。
2021年11月22日,来自梅奥医学中心/斯坦福大学Cornelia Weyand课题组
(第一作者为
吴博文
博士)
在Nature Immunology杂志上发表了文章
Mitochondrial aspartate regulates TNF biogenesis and autoimmune tissue inflammation
,
报道了功能失调的线粒体造成天冬氨酸穿梭缺陷驱动了类风湿性关节炎患者T细胞内质网 (ER) 扩张和TNF过度合成。
越来越多证据表明,自身免疫疾病中异常的免疫细胞代谢可促进炎症的细胞和分子过程。在 RA 患者的 T 细胞中,葡萄糖代谢从经典的糖酵解转移到磷酸戊糖途径,该途径通过产生烟酰胺腺嘌呤二核苷酸磷酸
(NADPH)
、戊糖和 5'-核糖磷酸来支持细胞合成代谢和增殖。同时,由 AMP 激酶
(
AMPK
)
活化丧失导致的线粒体功能缺陷,将 T 细胞代谢从脂肪酸氧化重定向到脂肪酸合成,以促进细胞因子的产生和组织浸润。除了作为代谢中心之外,线粒体还可通过钙和活性氧等调节广泛的信号通路来控制基因表达。有趣的是,正如本研究中报道的,线粒体还可以通过其他机制影响免疫细胞效应表型。
研究者首先确定了 RA 患者的 T 细胞线粒体去极化及功能失调,并且这种线粒体表型与 ER 的扩张有关。ER 是一种与核糖体偶联的膜结构,负责复杂跨膜蛋白的翻译。RA患者T细胞内ER含量明显高于正常对照,而且抑制线粒体活性可以模拟病理性的ER扩张。T细胞内ER的大小与TNF合成分泌直接相关,病人来源的T细胞显著上调TNF的产生,而抑制ER扩张可以显著改善这一特性。
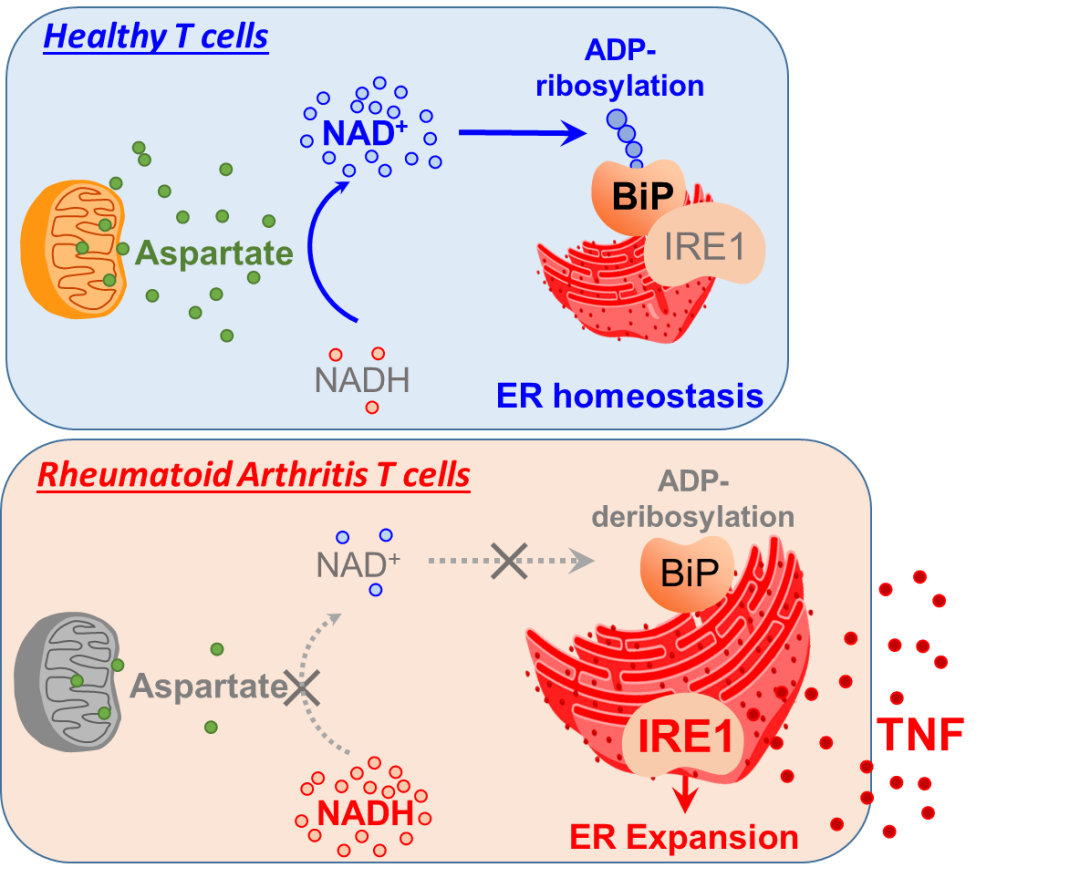
机制方面,由于氧化代谢改变,RA 患者来源的T 细胞线粒体中天冬氨酸
(一种源自草酰乙酸的氨基酸,三羧酸循环的中间体)
的丰度降低。天冬氨酸通常从线粒体穿梭到胞质,在那里被转化回草酰乙酸,参与细胞质烟酰胺腺嘌呤二核苷酸
(NAD+)
的再生。有缺陷的天冬氨酸穿梭导致NAD+ 的水平降低,减少NAD+依耐的蛋白质 ADP-核糖基化。ER 分子伴侣BiP在没有 ADP 核糖基化的情况下,可以释放内质网应激蛋白
(如 IREα)
,从而驱动内质网扩张并将 T 细胞转化为 TNF 超级生产者
(图一)
。有趣的是,用外源性 NAD+、天冬氨酸处理RA T 细胞可防止 ER 扩增。
图一:线粒体Aspartate通过调节BiP ADP-ribosylation进而控制内质网扩增及TNF合成示意图。
最后,该研究还证明,向RA患者T细胞中转染健康线粒体可抑制 ER 扩增并阻止 TNF 的合成。为了探索该研究的转化价值,作者使用了免疫功能低下的 NSG 小鼠,该品系缺乏 B 细胞、T 细胞和 NK 细胞。他们将人类滑膜组织移植到这些小鼠身上,随后注射 RA 患者的外周血单核细胞以诱导滑膜浸润并模拟人类RA疾病状态。当小鼠用天冬氨酸治疗时,滑膜 T 细胞浸润大大减少,同时产生几种促炎细胞因子,包括但不限于 TNF。
这项研究最显着的发现是线粒体功能与 ER 生物发生和 T 细胞产生 TNF 之间的耦合。目前针对 RA 和许多其他自身免疫性疾病患者的生物治疗主要通过抗体针对炎性细胞因子或其受体。最新的小分子抑制剂可通过抑制 Janus 激酶
(JAK)
抑制致炎细胞因子信号。所有这些策略都旨在阻止长期炎症过程中的下游事件。然而,细胞因子及其下游信号通路对免疫细胞外多种细胞都有广泛影响,这一事实与现有治疗手段的不良事件相关,例如接受 JAK 抑制剂治疗的患者的血管栓塞事件。
本研究提示了干预炎症过程上游生化和代谢事件可能具有临床应用潜力,免疫细胞中线粒体功能以及其对其他细胞器的调控具有相当重要的研究价值。
https://www.nature.com/articles/s41590-021-01065-2
1. McInnes, I. B. & Schett, G. N. Engl. J. Med. 365, 2205–2219 (2011).
2. Zhang, F. et al. Nat. Immunol. 20, 928–942 (2019).
3. Wu, B. et al. Nat. Immunol. https://doi.org/10.1038/s41590-021-01065-2 (2021).
【非原创文章】本文著作权归文章作者所有,欢迎个人转发分享,未经允许禁止转载,作者拥有所有法定权利,违者必究。


 个人中心
个人中心
 我是园区
我是园区
 退出
退出