

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!



什么是DLT
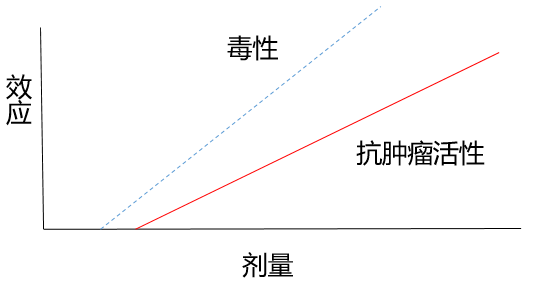
DLT的标准是什么
与试验药物有关的≥ 3级的非血液学毒性
与试验药物有关的≥ 4级的血液学毒性
在G-CSF(粒细胞集落刺激因子)出现之前,“≥ 3级中性粒细胞减少”就认为是DLT;在G-CSF出现之后,很多试验把 “≥ 4级中性粒细胞减少”才认为是DLT。
在使用酪氨酸激酶抑制剂(TKI)治疗后,经常会出现一过性的、无症状的3级血生化异常或高血压,不一定需要干预,或者通过口服降压药就可以缓解。这些3级毒性往往也不需要设定为DLT。
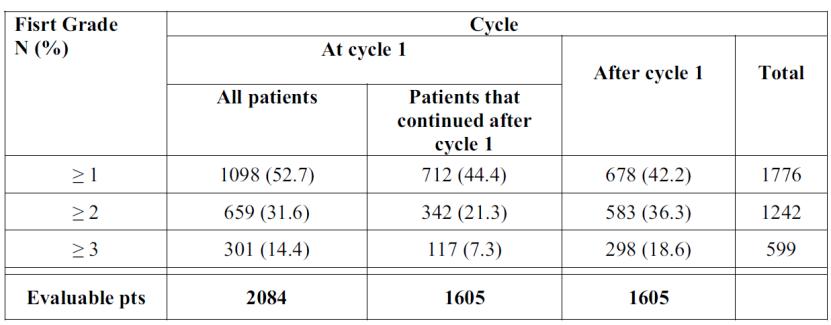
DLT评估期得设定多长时间?
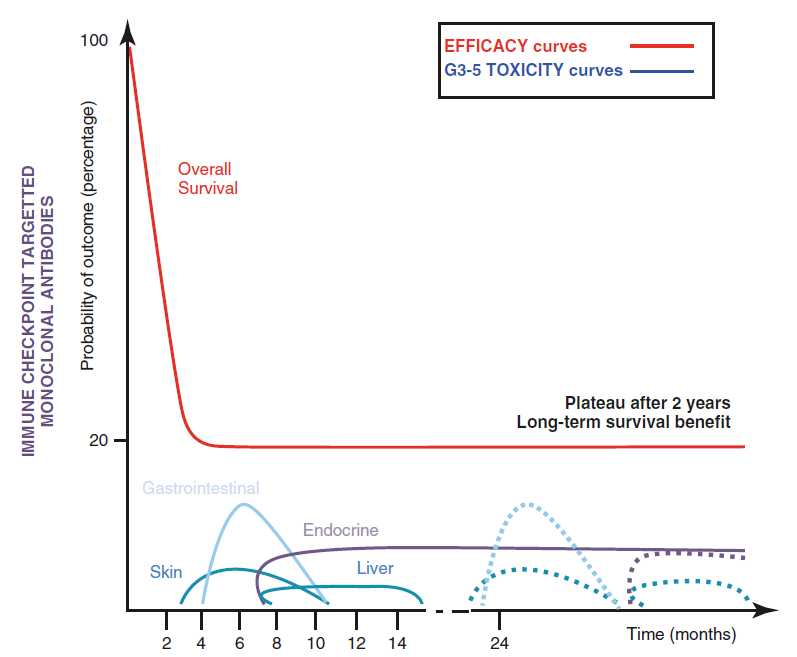
免疫检查点抑制剂的总生存(OS)曲线和3-5级严重免疫相关不良事件(irAEs)的概率
参考文献(上下动查看更多)
1. Postel-Vinay S, Collette L, Paoletti X, Rizzo E, Massard C, Olmos D, et al. Towards new methods for the determination of dose limiting toxicities and the assessment of the recommended dose for further studies of molecularly targeted agents--dose-Limiting Toxicity and Toxicity Assessment Recommendation Group for Early Trials of Targeted therapies, an European Organisation for Research and Treatment of Cancer-led study. Eur J Cancer. England; 2014;50:2040–9.
2. Le Tourneau C, Razak ARA, Gan HK, Pop S, Diéras V, Tresca P, et al. Heterogeneity in the definition of dose-limiting toxicity in phase I cancer clinical trials of molecularly targeted agents: a review of the literature. Eur J Cancer. England; 2011;47:1468–75.
3. Eisenhauer, Elizabeth A., Christopher Twelves, and Marc Buyse, eds. Phase I cancer clinical trials: a practical guide. Oxford University Press, 2015.
4. Yap, Timothy A., Jordi Rodon, and David S. Hong, eds. Phase I Oncology Drug Development. Springer International Publishing, 2020.
5. Lutzky, Jose, et al. "A phase 1 study of MEDI4736, an anti–PD-L1 antibody, in patients with advanced solid tumors." (2014): 3001-3001.
6. Wong, Han Hsi, and Sarah Halford. "Dose-limiting toxicity and maximum tolerated dose: still fit for purpose?." The Lancet. Oncology 16.13 (2015): 1287-1288.







