

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!
撰文 | 章台柳
责编 |兮
有研究表明体内肿瘤细胞的代谢与体外培养的癌细胞的代谢具有差异【1】。肿瘤微环境在一定程度上促成这种差异,肿瘤细胞经常因血管系统功能失调而缺乏营养和氧气;而培养基的环境营养成分和氧气的变化会对癌细胞的代谢产生广泛的影响。此外,实体瘤的环境非常复杂,包含多种细胞类型,如血管细胞、成纤维细胞和免疫细胞。许多非癌细胞类型不断相互作用,为癌细胞生长和逃避免疫监视提供代谢支持。这些观察结果都提出一种可能性,即在培养条件下所做的许多研究工作也许不能反映人类肿瘤细胞的复杂性。
胰腺导管腺癌(PDAC)是一种侵袭性肿瘤,具有致密的纤维增生性间质、严重缺氧和免疫抑制微环境等特征。KRAS和TP53突变较为常见,促进代谢变化以支持合成代谢过程和营养素的清除。之前的研究显示自噬、半胱氨酸摄取和丙氨酸代谢等代谢途径都参与PDAC的疾病进展【2-4】。然而,我们对胰腺肿瘤进展过程中的基本代谢途径或环境因素如何导致这些代谢依赖性缺乏完整的了解。
近日,Cell Metabolism杂志背靠背发表两篇文章Functional Genomics In Vivo Reveal Metabolic Dependencies of Pancreatic Cancer Cells和Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer,对胰腺导管癌细胞在体内和体外培养过程中代谢基因的依赖性进行筛选分析,发现两种条件下,PDAC细胞对大多数代谢基因的依赖性具有类似性,仅对一小部分代谢基因的依赖性具有差异。


第一篇是来自洛克菲勒大学Kıvanç Birsoy的文章,题目为Functional Genomics In Vivo Reveal Metabolic Dependencies of Pancreatic Cancer Cells。研究人员首先构建靶向2900个代谢基因的sgRNA库在小鼠胰腺癌细胞系(KrasG12D/Trp53R172H PDAC小鼠模型来源)中进行loss-of-function筛选。转染sgRNA文库后,将敲除细胞系传代培养并在免疫活性良好的B6小鼠中形成皮下肿瘤。随后对体外培养和体内生长的PDAC进行sgRNA分析,发现两者的sgRNA丰度具有显著的相关性,意味着即使两种生长条件下营养和细胞内信号的不同,胰腺癌细胞在体内的代谢本质有很大一部分是相似的,可能不是由肿瘤环境决定的。同时,筛选过程鉴定出一小部分代谢基因在两种条件下具有差异性的依赖性。其中,体内肿瘤依赖性的代谢基因在血红素代谢、氧化磷酸化、核苷酸合成和抗原递呈相关的通路上富集,同时涉及到自噬、脂质合成相关基因。
对比Kras驱动的胰腺癌细胞系和Kras驱动的肺癌细胞系对代谢基因的依赖性,发现两种细胞对代谢基因的依赖性重叠部分很少,表明组织来源是体内代谢依赖性的一个重要决定因素。值得注意的是,肺癌和胰腺癌中血红素合成基因(Uros、Cpox、Ppox和Hmbs)的sgRNA丰度都在体内生长条件中减少,表明无论来自何种组织,都存在一种普遍的环境压力,导致肿瘤对血红素合成具有代谢依赖性。为了验证这一假说,在Kras驱动的胰腺癌和肺癌细胞系中敲除Hmbs。Hmbs的缺失对细胞系的体外培养并没有较大的影响,显著减少体内肿瘤的大小。那么肿瘤环境哪些因素导致这种代谢依赖性?首先,观察到血红素分解代谢的限速酶——Hmox1,在肿瘤和缺氧条件下表达上调。那么,Hmox1的上调增加血红素的分解,可能会掩盖肿瘤生长对血红素的依赖性。进一步在Hmbs缺陷的胰腺癌细胞中敲除Hmox1,则部分逆转Hmbs缺失导致的肿瘤生长抑制。其次,利用sgRNA靶向血红素合成基因显著抑制KRAS突变的胰腺癌病人来源异种移植(PDX)的生长,而且将血红素合成基因作为一个基因特征对病人进行评分分析,发现较高的血红素合成基因与较低的无疾病生存期具有相关性。
是否存在部分的代谢基因参与肿瘤细胞的免疫逃逸?对免疫缺陷小鼠NSG进行筛选分析,其中参与自噬(macroautophagy)的关键酶Atg7在两者具有差异,这提示我们自噬或是免疫逃逸或免疫介导细胞死亡的关键过程。敲除Atg7导致免疫活性良好的B6中肿瘤显著变小,而在免疫缺陷的NSG小鼠(缺少T、B、NK)和Rag1-/-小鼠(缺少T、B)中这种差异消失。同时Atg7缺陷的肿瘤中caspase-3的活化增加,免疫介导的死亡增加,免疫细胞的浸润没有改变,但CD8+ T和NK表达IFN γ增加。进一步研究发现Atg7缺失导致肿瘤细胞对TNF α诱导的死亡更加敏感,即自噬可保护PDAC细胞免受免疫细胞如CD8+ T分泌的TNF α诱导的细胞死亡,从而帮助癌细胞实现免疫逃逸。

第二篇文章来自纽约大学医学中心的Alec C. Kimmelman和Dana-Farber癌症研究所的Andrew J. Aguirre合作发表,题目为Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer。研究人员通过对PDAC体内生长和体外培养条件下代谢基因的CRISPR-Cas9筛选,同样发现体内和体外条件下,癌细胞对代谢的依赖性具有惊人的相似性,同时肿瘤内细胞的营养共享可能掩盖代谢基因的依赖性(比如Hmox1上调掩盖了肿瘤细胞对的Hmbs依赖性)。
虽然肿瘤细胞在3D培养系统中增殖速率更慢,营养和氧气具有限制,但研究人员进一步证明3D培养系统能更好地模拟肿瘤代谢-细胞信号。例如,Fdft1编码甲戊酸途径中的一种酶,催化焦磷酸法尼酯合成角鲨烯,是胆固醇合成的第一步。2D、3D和体内肿瘤生长对Fdft1具有不同的依赖性,2D生长不需要Fdft1,但3D和体内增殖都需要Fdft1。Fdft1缺失导致PDAC细胞在3D和体内生长被抑制,过表达Fdft1则能改善Fdft1缺失导致的生长抑制。同时,相比于免疫缺陷小鼠,Fdft1缺失PDAC在免疫活性良好小鼠中生长更慢,说明Fdft1活性缺失增强其对适应性免疫的敏感性,并且免疫组化分析显示肿瘤中CD8+ T细胞的浸润增加。对细胞中信号通路进行分析,发现Fdft1缺失细胞中Akt、S6的磷酸化水平降低,Erbb3蛋白减少,限制PDAC细胞在3D培养条件下的生长。利用Fdft1抑制剂对肿瘤进行治疗,显著减少肿瘤的生长,增加肿瘤中CD8+ T细胞的浸润和肿瘤细胞的凋亡。此外,PDAC病人中,FDFT1的高表达与较差的总体生存率显著相关。FDFT1抑制或可作为治疗PDAC的一个潜在靶点。
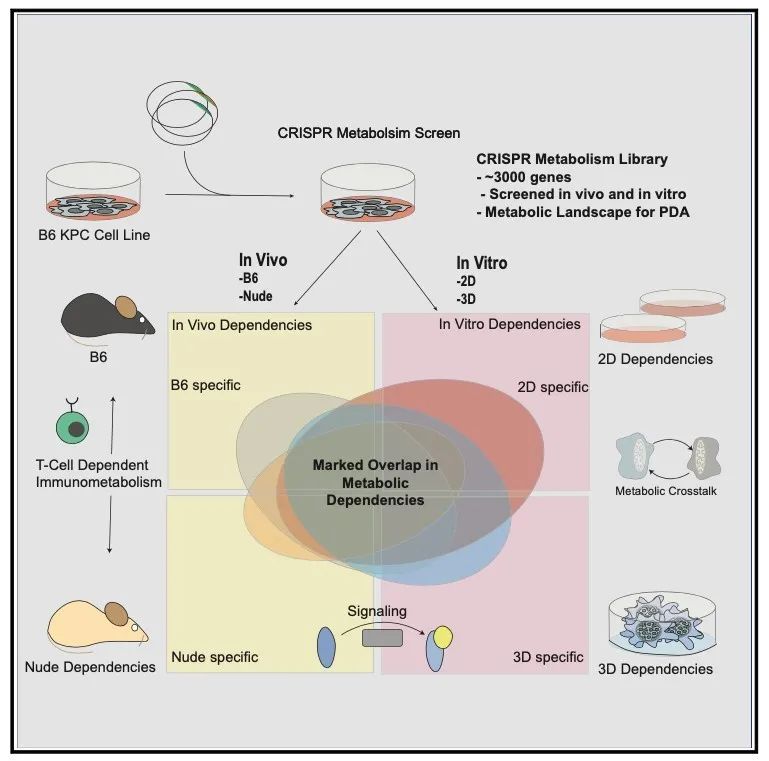
总的来说,两项研究同时聚焦胰腺癌细胞在体内和体外生长对代谢基因的依赖性,揭示出两种条件下对代谢基因的依赖性具有很大程度的相似性,同时又揭示出两者的不同,为研究肿瘤微环境限制和免疫系统的代谢依赖性提供了资源,同时为寻在潜在的抗癌靶点提供了有力的工具。
原文链接
https://doi.org/10.1016/j.cmet.2020.10.017
https://doi.org/10.1016/j.cmet.2020.10.018
制版人:琪酱
参考文献







