

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

在上篇,时岚博士介绍了抗肿瘤药物早期临床研究的传统设计,梳理了I期、II期临床试验设计的要点。众所周知,抗肿瘤药物的I、II期临床试验研究效率将对药物整体的研发进度和成功率起到举足轻重的作用。因此,有必要在早期开发和探索阶段寻找新的方法和新思路,从而缩短药物的研发时间并降低研发成本。在下篇,时岚博士将介绍包括无缝设计、篮式试验和伞式试验等在内的抗肿瘤药物早期临床研究设计的新方法。

适应性设计
“无缝”设计
传统的药物早期临床开发阶段往往分为I期和II期,尽管I期临床试验的主要目的是探索药物的MTD,但同时也会累积不少有关药物疗效的信息,而这些信息也应当被II期临床试验所采用。因此I/II期无缝设计(phase I/II seamless design)是一种更为有效的设计,可最大程度地利用I期临床试验中药物的安全性和疗效方面的信息,实现I期和II期的整体化协同研究,减少试验的样本量,缩短临床试验的研究时间,最终筛选出最优的药物剂量用于后续确证性的III期临床试验。
早在2006年,FDA发布的《关键性通道机遇目录》(Critical Path Opportunities List,CPOL),就鼓励适应性设计。2018年9月28日,FDA发布了《药品和生物制品临床试验的适应性设计》(Adaptive Designs for Clinical Trials of Drugs and Biologics)和《主研究方案:以促进抗肿瘤药物和生物制品研发为目标的高效临床试验设计策略》(Master Protocols: Efficient Clinical Trial Design Strategies to Expedite Development of Oncology Drugs and Biologics)两份指南,重点讨论了临床试验适应性设计和肿瘤药物临床试验的主研究方案。
无缝试验的研究方式使得传统的各个研究阶段不再泾渭分明,取而代之的是临床药理学、探索性研究和验证性研究之间评估的相互重叠。无缝试验设计下,不仅药物在研发早期就会对疗效进行评估,还可以使用更少样本量的患者探索不同疾病领域药物的疗效,这使得研发效率得以大大提高。截至2017年9月,FDA已批准40多个新药临床研究申请(Investigational New Drug,IND)在首次人体试验中应用无缝试验设计。
以奥希替尼为例,奥希替尼是FDA有史以来获批上市速度最快的抗癌药物。通常,抗癌药物从开始进入临床试验阶段直到被FDA批准上市,平均需要花费10年以上的时间,而奥希替尼仅仅用了2年半。奥希替尼在FDA的快速获批,得益于其在I期到II期的无缝试验设计,大大节省了研发周期。凭借奥希替尼出色的临床试验结果加之其合理的适应性设计,FDA授予其多种审评“绿色通道”,最终其以2项关键性II期单臂临床试验的结果获得了第一个适应证的上市许可。

图 1 I/IIa/IIb期无缝设计—奥希替尼
时岚博士提醒大家,在进行I/II期无缝设计时,需要考虑的问题有:1)进行人群扩展是否有合理的理由;2)样本量是否符合既定目标和终点;3)是否有适当的统计计划适用于已定终点;4)入组标准是否适合扩展组;5)对于药物显示出有效或无效,试验是否有明确的结束终点;6)是否有一个适当的系统用于及时跟所有研究者沟通;7)是否充分探索和了解试验药或者同类药的安全性和有效性最新信息;8)如果该试验用于注册申请,是否设立独立评审委委员会(Independent Review Committee,IRC),是否提前与监管机构进行沟通;9)临床前试验结果(特别是长毒试验)是否支持I/II期无缝试验;10)如果要考察食物影响,那么食物影响试验涵盖在I/II期无缝试验中还是单独开展一个I期试验来探索。
篮式试验
篮式试验(Basket Trial)即一项试验包含多种适应症,将所有相同驱动基因突变、不同肿瘤来源的患者放在一个“篮子”进行的研究。事实上,篮式试验正是为了满足靶向药物的研发应运而生,由于其主要关注相应的分子变异,而不限制肿瘤组织学来源和分型,因此为发病率较低、临床试验中入组困难的罕见肿瘤的药物研发提供了可能。对于药物研发企业,篮式试验可实现在较少样本量的情况下,同时研究一种药物对多种肿瘤类型的治疗效果,可以显著缩短药物上市的时间,从而减少研发成本。此外,向美国食品药品监督管理局(Food and Drug Administration,FDA)、中国国家药品监督管理局(National Medical Products Administration,NMPA)等药品审评部门提交注册申请时,II期篮式试验的研究结果也可为申请药物的疗效和安全性提供依据。
以维罗非尼为例,BRAF突变可以在多发性骨髓瘤、黑色素瘤、卵巢癌、结肠癌、甲状腺癌、绒毛膜癌、胃肠肿瘤、肺癌等多个癌种中被检出。因此,首个篮式试验——维罗非尼的临床研究中,入组了多个瘤种的患者。
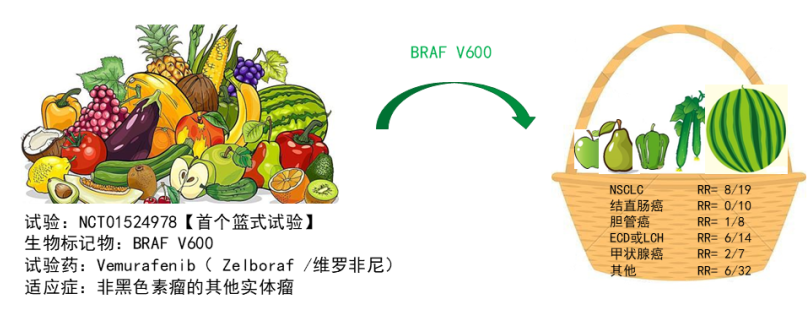
图 2篮式试验—维罗非尼
伞式试验
伞式试验(Umbrella Trial)即一项试验中评估多种产品,将不同的靶点基因检测在同一时间里完成,然后根据不同的靶基因分配相应的精准靶向药物治疗的研究。以肺癌为例,把KRAS、EGFR、ALK等不同的驱动基因聚在同一把雨伞下进行研究。“伞式试验”的优势是将非常少见的突变事件集中起来,变少见事件为“常见”事件,既有助于少见疾病临床试验的开展,也有助于患者获得精准治疗的机会。
 图 3 伞式试验—肺癌
图 3 伞式试验—肺癌
伞式设计旨在评估多种药物针对同一种疾病或生物标记物类型的靶向治疗的临床效果,该设计在一个整体临床试验方案中含有多个子方案,每一个子方案针对一种或多种药物,可能是单臂试验或随机对照试验。伞式试验常用来为确证性研究选择候选药物,也可用来作为确证性研究。
单臂试验设计
单臂临床试验设计即在临床试验设计时,不设立平行对照组,而仅采用外部对照(包括历史对照),将接受试药物的一组患者与该研究以外的一组患者的结果进行比较。单臂试验结果用于注册申请,是一种附条件加上批注上市的策略。
对于临床急需,具有治疗优势,安全性风险可控的药物,可基于I/II临床研究数据有条件批准上市,后续开展确证性III期研究。监管机构的共性要求为:1)在FAS集(Full Analysis set)评价有效性,独立评审委员会(Independent Review Committee,IRC)评价的客观缓解率(Objective Response rate,ORR)兼顾缓解持续时间(Duration of Response,DOR);2)安全性暴露量:不得少于300例(RP2D剂量及以上);3)最快申报时间:末例受试者接受治疗并完成至少两次影像学评价后;4)新药申请(New Drug Application,NDA)审评期间滚动递交DOR、无进展生存期(Progression Free Survival,PFS)、总生存期(Overall Survival,OS)等数据。
单臂研究的优势有:1)样本量少;2)随访时间短;3)新药研发成本低;4)与随机对照试验(Randomized Controlled Trial,RCT)相比,单臂设计将临床研发时间缩短了约2年(基于FDA数据)。但是,单臂研究也存在不足。首先,历史对照具有局限性:1)研究人群存在相关差异:性别、年龄、组织病理分型、分子分型、疾病分期、ECOG、既往治疗等;2)研究时期相关差异:不同时期患者的预后不同(例如,对照治疗方案的改良;后续治疗差异影响OS;支持治疗差异),诊断标准、疾病分期方法的改良(例如,乳腺癌的分子病理权重增加),肿瘤评估仪器及标准的改良;3)研究地区相关差异:人种、病因、发病机理、病理学、流行病学、前期治疗标准、现有治疗的有效性。其次,对于抗肿瘤药,ORR并不一定能够预测PFS、OS、疾病进展时间(Time To Progress,TTP),因为不同类型肿瘤的自然进程变异性很大,ORR不一定能充分体现时间-事件终点。
有关FDA对单臂结果的认可度,在2005年之前,FDA加速审批的抗肿瘤药产品中,占总数1/3的品种是基于RCT数据,占总数2/3的品种是基于早期的单臂数据。2005年之后,FDA更倾向于采用RCT的数据,获批的申请中,随机对照研究和单臂的比例为1:1。在获批的47个适应症中:26个被后续的研究证实了临床获益转为常规批准(55.3%);3个(6%)被证明无效而撤回;14个(29.7%)未完成确证性试验;21个因未证实临床获益而未能转为常规批准(8个使用了替代终点指标的RCT,13个为单臂)。
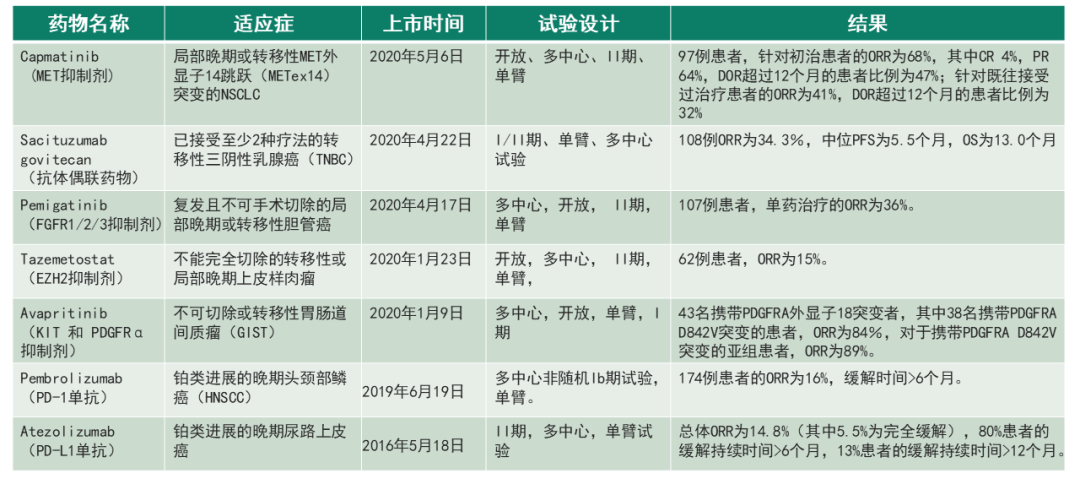
表 1国外依据单臂试验批准上市的案例
单臂试验在国内认可度,根据《药品注册管理办法》第十三条:国家药品监督管理局建立药品加快上市注册制度,支持以临床价值为导向的药物创新。对符合条件的药品注册申请,申请人可以申请适用突破性治疗药物、附条件批准、优先审评审批及特别审批程序。在药品研制和注册过程中,药品监督管理部门及其专业技术机构给予必要的技术指导、沟通交流、优先配置资源、缩短审评时限等政策和技术支持。
根据《单臂试验支持注册的抗肿瘤创新药进入关键试验前临床方面沟通交流技术指导原则》:申请人在药物临床试验申请前、药物临床试验过程中以及药品上市许可申请前等关键阶段,可以就重大问题与药品审评中心等专业技术机构进行沟通交流。为切实鼓励创新,保障抗肿瘤创新药以充分科学依据开展关键单臂试验,帮助申请人提高研发效率并与中心更高效地沟通,制定本指导原则,以期为计划以单臂试验支持注册的抗肿瘤创新药进入关键试验前临床方面沟通交流提供资料准备建议和技术指导。
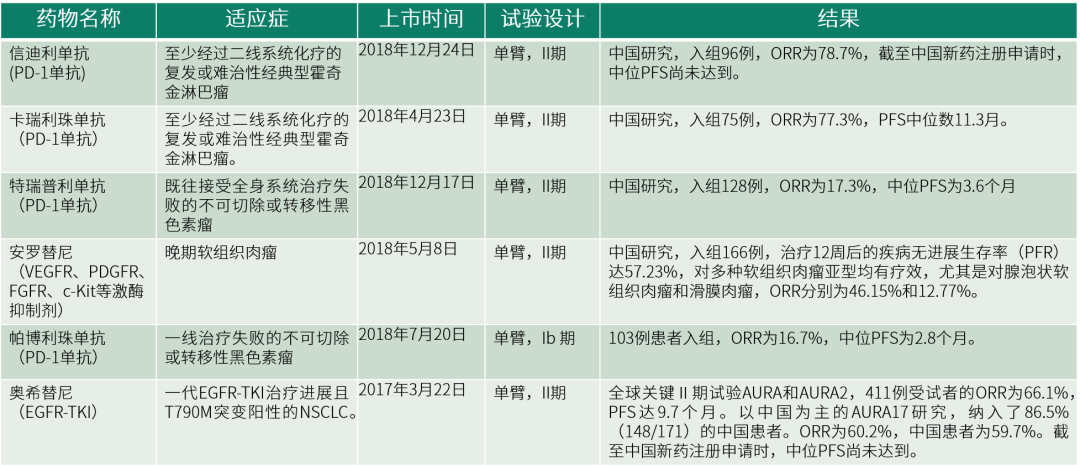

表 2国内依据单臂试验批准上市的案例
目前,新药在美国可通过FDA的优先审评、加速审批、快速通道和突破性疗法4条途径来加快审评速度。2019年,NMPA为我国鼓励创新和满足临床急需而设立了包括突破性治疗、附条件批准、优先审评审批、特别审批在内的四个加快通道。时岚博士建议,申办者应在临床试验申请前及上市许可申请前两个关键的时间节点与CDE密切沟通,尤其是在临床试验申请前,可就单臂试验设计的意向,与CDE审评老师积极沟通。
扩展阅读:
关于泰格医药科学事务部
科学事务部团队专注于I-IV期药物和医疗器械临床试验方案、BE试验方案、临床研究总结报告、新药IND和NDA申报资料、临床开发计划、临床研究综述、研究者手册、药物临床试验风险控制计划和患者知情同意书等医学相关资料的撰写。成立十余年来,为国内外知名制药企业、创新药研发公司提供优质、高效、合规的医学撰写服务,已经累积了数百个临床项目的撰写经验。项目经验涉及各个疾病领域,尤其擅长肿瘤、呼吸、免疫、神经、血液、消化、骨伤等;涵盖的产品以创新药为主,也有仿制药和医疗器械,更有当下肿瘤药物研究领域的热点研究药物。目前,科学事务部团队由近30位具备医药背景的高学历人才组成,核心成员具备丰富的医学资料的撰写和翻译经验,可与其他职能部门密切合作,共同保障临床试验和质量和进度。
文 | 任倩倩
Tigermed
泰格医药
泰格医药(股票代码:300347.SZ/3347.HK)是行业领先的一体化生物医药研发服务平台,为全球制药和医疗器械行业提供跨越全周期的临床研究创新解决方案。通过全面的服务体系和顶尖的质量标准,我们助力生物医药产业提升研发效率、降低研发风险,确保研究项目高质量交付,加速医药产品市场化进程,履行对行业和患者的承诺。同时,我们也通过覆盖各领域的60多家子公司,打造赋能全产业链的创新生态,推动医疗产业创新和发展。作为全球化的研发平台,泰格医药在全球布局150多个办事处和研发基地,拥有超过7000人的专业团队,覆盖5大洲的39个国家,致力于解决最具挑战的全球健康问题,满足患者的未尽医疗需求,创造社会价值,造福人类健康。
网址: www.tigermedgrp.com
邮箱: marketing@tigermedgrp.com
注:本公众号发布内容仅供一般参考之用,不可视为详尽说明,亦不构成信息披露和投资建议,投资者不应以该等内容取代其独立判断或仅根据该等内容做出决策。








