

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

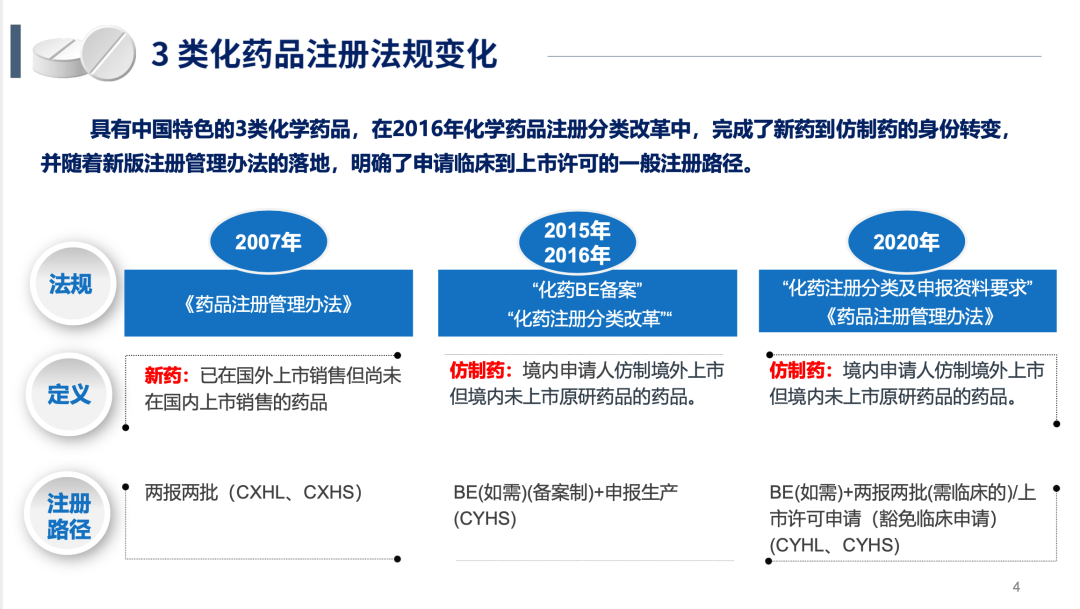
“新3类化药”可能的注册情形
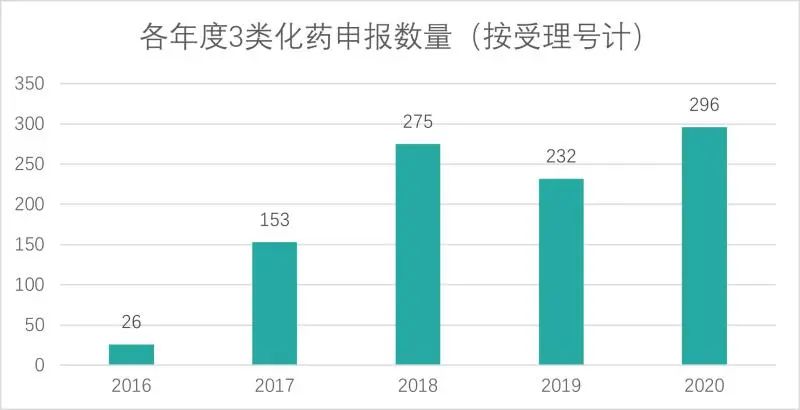

朱林和谢昵总结了3类化药可能需要进行的注册临床试验情形:
第一种是BE试验,可豁免验证性临床试验仅以BE试验申请注册;
第二种是豁免BE及其他临床试验,由境内外临床数据支持,通过体外评价证实与原研一致;
第三种是验证性临床试验,无法或无需进行BE试验的品种,通过验证性临床试验证实有效及安全性;
第四种是BE+验证性临床试验,BE证实与原研生物等效,验证性临床确证疗效及安全性;
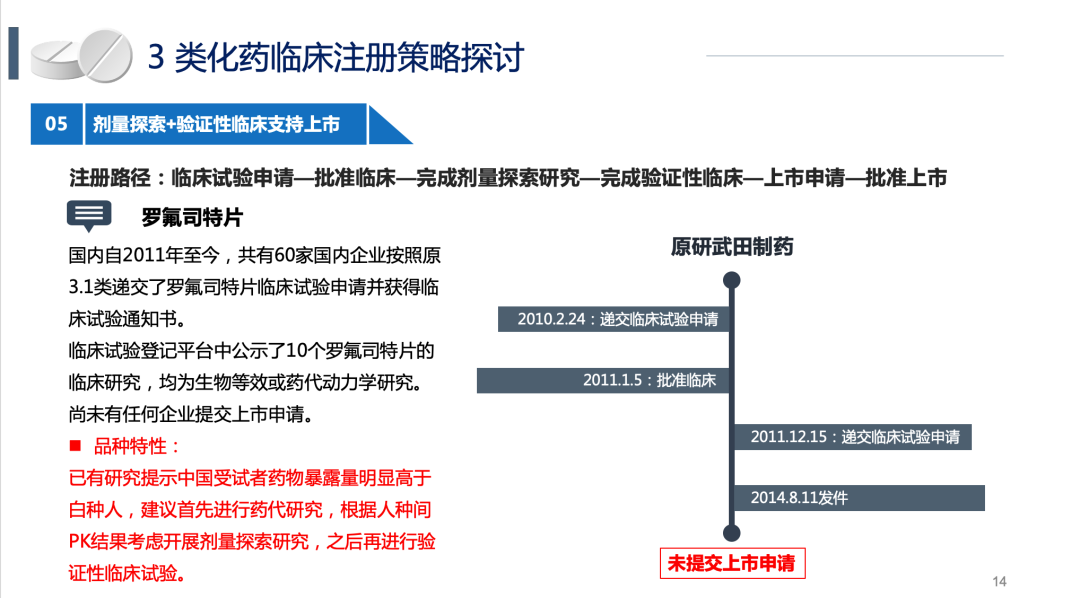
第五种是剂量探索+确证性临床研究,有种族差异的,需以新药临床思路进行仿制药临床研究。
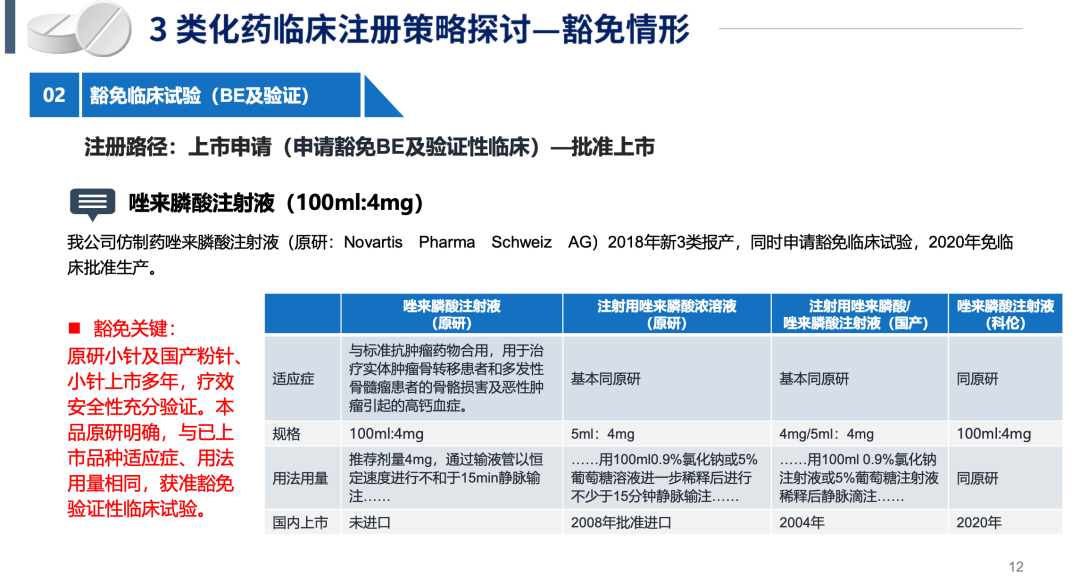
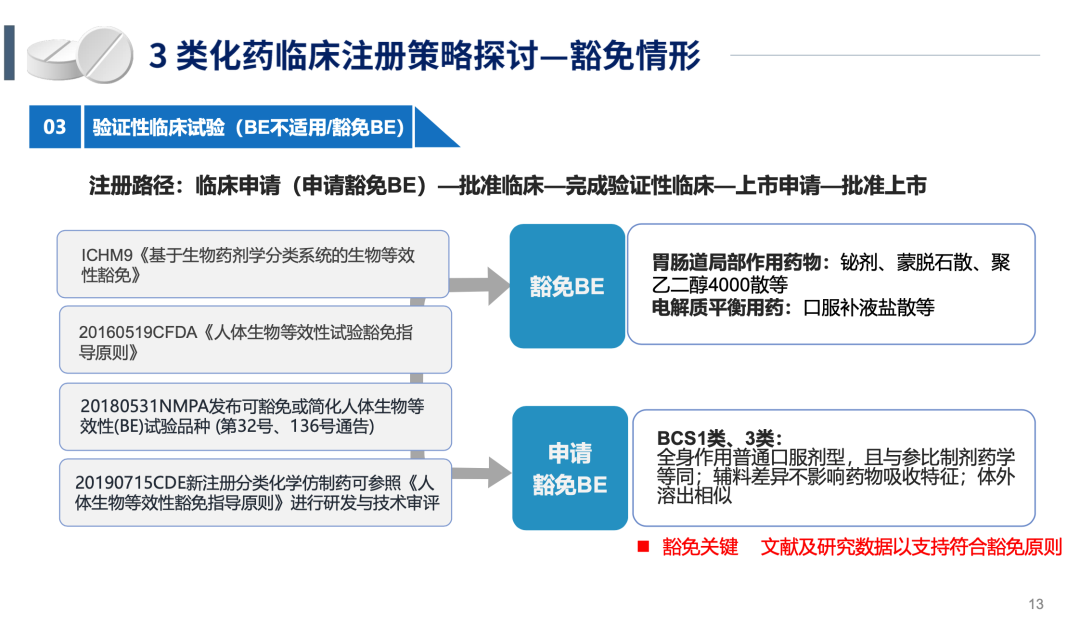
BE试验和豁免临床的具体案例

互动

嘉宾
常建青 泰格医药政策法规事务副总裁
付洁鹰 应世生物注册事务高级总监
寇壬花 百济神州药政科学部高级总监
桑艳双 百济神州注册副总监
朱林 恒瑞医药注册事务总监

(感谢谢昵老师和其他讲者对本文的指导)







