

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!
作者:林出
标的公司:BioMarin Pharmaceutical Inc. (NASDAQ:BMRN)
BioMarin Pharmaceutical Inc.(BMRN)宣布,其正在进行的全球III期Gene8(GNXE-OLD-1) 研究,指征成人严重血友病A的研究性基因疗法的积极结果。这是迄今为止针对任何适应症的任何基因疗法的最大的全球III期研究,有134名参与者。研究的所有参与者都接受了单剂量的valoctocogene roxaparvovec,并完成了一年/+的随访。
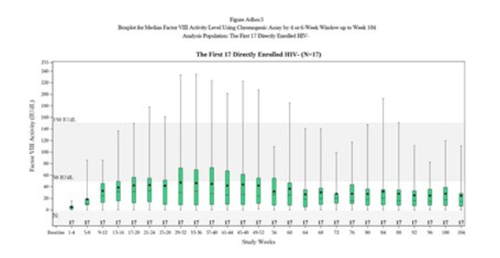
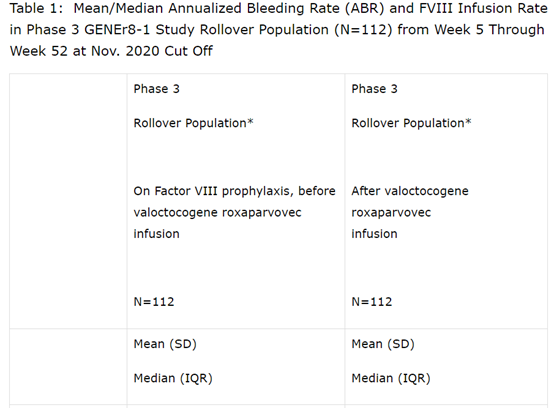
III期GENEr8-1研究(平均随访71.6周)的数据显示,在pre-specified的年化出血率(ABR)初步分析中,单剂量valoctocogene roxaparvovec可显著降低ABR,从基线时预期收集的4.8次(中位数2.8次)降至每年0.8次(中位数0.0次)(p值<0.0001)。80%的患者在治疗后第五周开始无出血。
Valoctocogene roxaparvovec还显著降低99%滚动人群中的平均年化Factor VIII,从135.9(中位数128.6)降低到每年2.0(中位数0.0)次(p值<0.0001)。
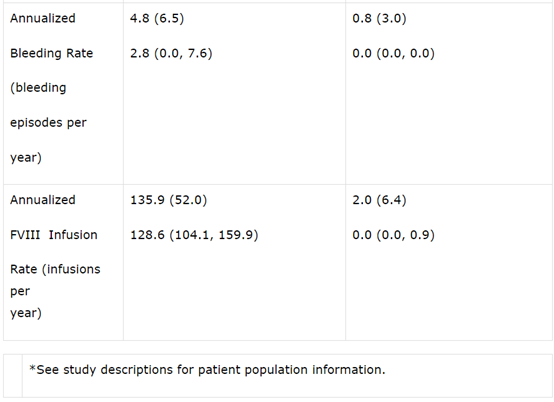
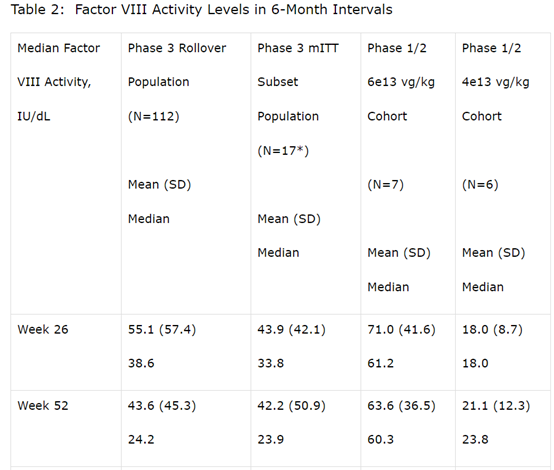
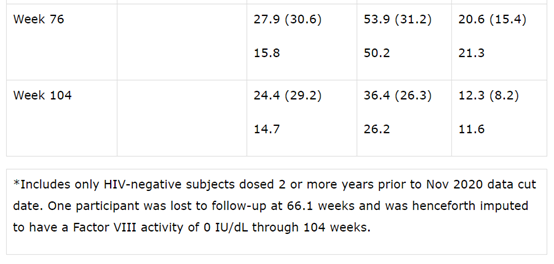
经发色底物(CS)测定,在输注valoctocogene roxaparvovec后第一年末,改良意向治疗(mITT)人群(N=132)的患者的内源性Factor VIII表达水平平均为42.9 IU/dL(SD 45.5,中位数23.9。与I/II期研究相比,Factor VIII表达下降的速度较慢,并保持在一定范围内,以提供止血效果。在数据截止日期前,至少两年服用mITT的人群中(N=17),Factor VIII的表达从第一年末的平均值42.2 IU/dL(SD 50.9,中位数23.9)下降到平均值24.4 IU/dL(SD 29.2,第二年末平均14.7),平均ABR为每年0.9次(平均0.0次)出血,显示出持续的止血效果。
Valoctocogene Roxaparvovec安全性
III期研究中,134名接受单次6e13 vg/kg剂量的患者对valoctocogene roxaparvovec的耐受性良好。没有参与者出现Factor VIII抑制剂或血栓栓塞事件。一名患退出未跟进。通过控制输液速率,输液相关反应得到有效缓解。
丙氨酸氨基转移酶(ALT)升高(N=115,86%)(肝功能检查),仍然是最常见的不良事件(AE)。其他常见的不良反应包括头痛(N=51,38%)、恶心(N=50,37%)、天冬氨酸转氨酶(AST)升高(N=47,35%)、关节痛(N=38,28%)和疲劳(N=37,27%)。22名(16.4%)患者共有43次严重不良事件(SAEs),所有SAEs均得到解决。
暂时使用皮质类固醇(或替代性免疫抑制剂)来控制ALT升高可能会出现常见的类固醇相关副作用。这些副作用的强度一般为1/2级,可控且可逆。长期高剂量皮质类固醇给药可观察到单独的3级类固醇相关副作用(如糖尿病、高血压、体重增加、骨折)。糖皮质激素相关的3级严重不良事件是一个安全问题,长期使用糖皮质激素是可逆的,只有体重增加。
总的来说,在I/II期研究中,valoctocogene roxaparvovec的安全性与先前报道的数据一致,没有延迟起效的治疗相关事件。没有患者出现Factor VIII抑制剂,也没有参与者退出研究。没有参与者出现血栓性事件。与valoctocogene roxaparvovec相关的最常见不良事件发生较早,包括短暂的输注相关反应和肝功能测试中测定的某些蛋白质和酶水平的短暂、无症状和轻度至中度升高,没有长期的临床后遗症。
GENEr8-1 Study Description
全球III期GENEr8-1研究评估了6e13 vg/kg剂量的valoctocogene roxaparvovec与目前的标准治疗FVIII预防性治疗相比的优越性。所有研究患者在基线时都有严重的血友病A,定义为FVIII活性≤1 IU/dL。这项研究共包括134名患者,所有患者在数据截取时都有至少12个月的随访。前22名患者直接进入III期研究,其中17名为HIV阴性,并且在数据截取日期前至少2年服用(referred to as the subset)。其余112名患者(滚动人群)在一项单独的非介入性研究中至少完成了6个月,以前瞻性评估出血事件,FVIII的使用,在GENEr8-1研究中,接受FVIII预防治疗后,再接受单次输注valoctocogene roxaparvovec治疗,与健康相关的生活质量。
I/II期剂量递增研究
I/II期剂量递增研究正在进行中,并继续对参与者进行长期监测。在这项研究中,共有15名严重血友病A和Factor VIII活性水平≤1 IU/dL的患者接受单剂量BMN 270治疗,其中7人接受6e13 vg/kg剂量的治疗,6人接受4e13 vg/kg剂量的较低剂量的治疗。另外两名受试者作为研究中剂量递增的一部分接受了较低剂量的治疗,但没有达到治疗效果。
监管状况
BioMarin正在与美国食品和药物管理局(FDA)合作,以取得针对严重血友病A的valoctocogene roxaparvovec基因治疗的市场批准。FDA建议该公司完成III期研究,并提交所有研究患者的两年随访安全性和有效性数据。此外,欧洲药品管理局(EMA)要求提供完整的III期研究的一年结果,以告知他们的收益风险评估。为便于在EMA regulatory framework内提交,BioMarin撤销了MAA,并计划在与该机构讨论后,于2021年第二季度向EMA重新提交这些数据。
FDA已经批准了valoctocogene roxaparvovec的突破性治疗方案。BioMarin的valoctocogene roxaparvovec已获得FDA和EMA的孤儿药资格,用于治疗严重血友病A。
Laurence A.Boxer,MD, 密歇根大学病理学密歇根医学部儿科和传染病研究教授,III期研究的研究员,“这是第一个在基因治疗试验中证明ABR优越性的统计证据。这些数据使我们对这一突破性的替代现有疗法充满信心,并使我们更接近于一种潜在的新治疗选择,以满足血友病A患者未得到满足的医疗需求,这一数据集增加了越来越多的科学和临床数据,为血友病A的valoctocogene roxaparvovec基因治疗创造了可能。”
Hank Fuchs, M.D.,BioMarin全球研发总裁,“在过去的七年里,我们进行了严格的科学研究和临床项目,以解决严重血友病A患者未得到满足的医疗需求,我们不断致力于推进这一有希望的研究性基因疗法治疗严重血友病A。我们对这些数据感到非常鼓舞,并期待着与监管机构合作,治疗医生,以及血友病A患者进一步了解这种基因治疗的潜力。”






