

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!
摘要
宿主细胞残留DNA是指可能出现于生物制品中的来自宿主细胞的DNA片段。用传代细胞株生产的生物制品,其宿主细胞残留DNA可能会传递肿瘤或病毒相关基因,存在潜在的危险性。各国药品监督管理机构对宿主细胞DNA的残留量有着严格的限度控制,同时各国药典也提供数种经典的检测方法。建立合适的宿主细胞残留DNA检测方法有助于监测生产工艺,确保生物制品的安全性和质量。此文对宿主细胞残留DNA潜在的危害、国内外监管机构对宿主细胞残留DNA检测标准和检测方法以及各方法的特点及研究进展做一综述。

前言
宿主细胞残留DNA是指可能出现于生物制品中的来自宿主组织细胞的DNA片段或更长的分子[1]。病毒性疫苗中宿主细胞残留DNA相关的风险主要是感染性和致癌性,如果存在逆转录病毒前病毒,其基因整合入受者基因组可能导致感染;如果存在病毒或传代细胞基质中的致癌基因,则可能具有引发肿瘤的潜在风险[2]。因此,建立合适的宿主细胞残留DNA的检测方法对监测生物制品的安全性和质量可控性至关重要。过去数十年,国内外各类监管机构出台了生物制品中可接受的宿主细胞残留DNA水平的指导方针。同时,各国药典也陆续提供了数种检测宿主细胞残留DNA的经典方法,包括阈值法、杂交法及实时定量PCR,但是传统的检测方法存在有弊端,目前以实时定量PCR为最优,其专一性强、灵敏度高、快速且可实现高通量检测[3]。
1
宿主细胞
病毒的生长和复制需要依存于活体生物或生物的组织与细胞,用于体外培养病毒的细胞统称为细胞基质。细胞作为疫苗等生物制品生产的主要原材料,其种类、代次、生长特性等将直接影响产品的质量和产量,尤其是安全性。目前,用于疫苗等生物制品生产的动物细胞基质有多种细胞,例如原代细胞、传代细胞、人二倍体细胞等。
1.1 原代细胞
原代细胞包括地鼠肾细胞、沙鼠肾细胞、牛肾细胞、猴肾细胞、鸡胚成纤维细胞等,主要用于减毒活疫苗和灭活疫苗生产。迄今为止,在我国已经批准生产的有地鼠肾细胞生产的狂犬病疫苗、乙型脑炎疫苗、双价肾综合征出血热灭活疫苗,沙鼠肾细胞生产的双价肾综合征出血热灭活疫苗,鸡胚成纤维细胞生产的麻疹减毒活疫苗等。如果细胞来源于无特定病原体动物,则受外源因子污染概率较低,一般不存在致癌基因和致癌蛋白,故致肿瘤的可能性极低。
1.2 传代细胞
传代细胞是指能够无限分裂繁殖传代培养的细胞系,常用的传代细胞有Madin-Darby犬肾(Madin-Darby canine kidney,MDCK)细胞、中国仓鼠卵巢(Chinese hamster ovary cell,CHO)细胞、非洲绿猴肾(Vero)细胞等。欧洲药典已经批准使用MDCK细胞生产流感疫苗。目前,此细胞系仅用于流感灭活疫苗的生产,有证据表明,培养基成分可以影响MDCK细胞是否具有致瘤性,尤其是无血清培养基[4]。CHO细胞被广泛用于表达重组DNA蛋白制品和重组乙型肝炎疫苗。有研究表明,CHO细胞的逆转录酶实验为阳性,具有致瘤的风险[5]。Vero细胞主要用于生产狂犬病灭活疫苗、乙型脑炎灭活疫苗、肠道病毒71型灭活疫苗、双价肾综合征出血热灭活疫苗等。美国FDA研究显示超过162代的Vero细胞致肿瘤实验为阳性,因此,大部分疫苗生产企业使用的细胞代次上限为130或140[6]。人二倍体细胞是指来源于正常人体组织且能在体外进行培养的细胞。人二倍体细胞虽然比其他传代细胞安全性更佳,但是由于其分裂生长速度慢,对于某些病毒不敏感,对这些病毒的培养感染滴度或产量明显低于其他细胞[7]。在国内建立并且得到批准用于疫苗生产的人二倍体细胞有2BS、KMB17细胞等,已经用于肠道病毒71型疫苗、甲型肝炎灭活疫苗、脊髓灰质炎减毒活疫苗、风疹减毒活疫苗等[8]。
2
残留DNA
生产生物制品的宿主细胞不尽相同,宿主细胞残留DNA带来的潜在危害也不同。这些DNA尽管有着相同的基本结构单位,但其片段长度不同,以不同的物理形式存在,进入人体后可造成多种多样的后果。
2.1 致癌性
残留DNA的主要致癌机制是引入了显性致癌基因,如MYC、RAS,这些显性致癌基因可直接转化正常细胞,使得部分细胞分化为瘤性细胞。其他致癌机制可能是宿主细胞残留DNA的插入引起突变,由于小片段的DNA整合到接种者基因组的概率较低,因此这种情况发生率也较低,即使恰巧在关键位置发生整合,激活一个原癌基因或抑制一个抑癌基因的概率也相应较低[9-11]。连续传代细胞的生长调控基因失调,理论上传代细胞系的DNA具有使其他细胞生长失控、产生致癌活性的潜在能力,因此需要对其DNA残留量进行质量控制。
2.2 感染性
Sheng-Fowler等[12]对于宿主细胞残留DNA的感染性进行了研究,他们用HIV-1前病毒DNA作为转染剂,建立了转染/共培养系统,首先对克隆前病毒DNA感染性进行定量测定,再对质粒进行酶切,转染293T细胞后,比较环状和线状DNA的量;通过测定逆转录酶活性确定感染性。结果表明,线状DNA的感染力是环状DNA的30~100倍,且2 mg残留DNA就具有感染性。少量的逆转录病毒DNA在体外是有感染性的,存在于细胞DNA中的前病毒与克隆的前病毒DNA具有同等的感染力,因为每个病毒感染所需的DNA量与两个基因组(病毒与细胞)的相对大小成比例。基于这些结果,在开发连续细胞系衍生生物制品时,需要考虑DNA的感染性以评估宿主细胞残留DNA的安全性。
3
国内外对残留DNA的限定标准
3.1 国内对残留DNA的限定标准
中国药典2020年版三部规定,以细胞基质生产的生物制剂DNA残留量不能超过100 pg/剂,以细菌或真菌基质生产的疫苗DNA残留量不能超过10 ng/剂。通则3407中介绍了3种外源性DNA残留量测定的方法,分别为DNA探针杂交法、荧光染色法和定量PCR法[13]。表1为我国目前检测DNA残留量的部分生物制品及标准汇总。

3.2 国外对残留DNA的限定标准
目前WHO、美国FDA和欧洲药典建议,进行风险评估时应考虑到细胞基质的特性及生物制品的用途。当使用具有致瘤性细胞系时,需要更加严格控制DNA残留量来确保产品安全性[14]。在一些产品生产过程中,很难在不改变产物效力的情况下降低DNA残留量,DNA烷化剂如β-丙內酯可以用于改变残留DNA的结构和大小,从而降低相关风险。
WHO和美国FDA现行指导方针推荐成品中残留DNA不高于10 ng/剂,长度不大于200 bp。美国FDA发布的指导原则中指出生物制品宿主细胞DNA残余限度为100 pg/剂,对于大剂量生物制品如单克隆抗体,根据其残留DNA来源及给药途径,DNA残留量可放宽至10 ng/剂[15]。
欧洲药典通则规定的生物制品残留DNA限度大多为不超过10 ng/剂,但对个别疫苗的残留DNA限定标准更严格,如甲型肝炎灭活疫苗中的DNA残留量不得超过100 pg/剂,乙型肝炎疫苗中的DNA残留量不得超过10 pg/剂[16]。
4
宿主细胞残留DNA定量检测方法
考虑到宿主细胞残留DNA的潜在安全风险,外源DNA还关系到产品的质量,建立灵敏、准确、特异性强、耐用性好的残留DNA定量测定方法十分重要[17]。尽管可以使用适当的方法在整个生产过程的各个阶段进行残留DNA分析,但没有普遍适用的方法来定量。中国药典2020年版附录收录的宿主细胞DNA残留量检测方法为DNA探针杂交法、荧光染色法和定量PCR法,美国药典2017年版USP40-NF35通则1130介绍了3种外源性DNA残留量测定的方法,分别为DNA探针杂交法、阈值法和实时定量PCR法[18],欧洲药典提出了2种定量宿主细胞残留DNA的灵敏分析方法,实时定量PCR和免疫酶法,并对这2种方法进行了对比[16]。
4.1 DNA探针杂交法
将供试品中的外源DNA加热变性为单链后吸附于固相膜上,在一定温度下与特异性标记的单链DNA探针杂交,两条单链DNA杂交复性成双链DNA,使用与特异标记物相应的显示系统显示杂交结果,与已知含量的阳性DNA对照比对后,显色深度反应DNA量,可测定供试品中外源性DNA残留量。然而,15个实验室用该方法在检测12个不同DNA含量的样品时,当样品DNA含量小于10 pg时,样品中其他化学物质对检测结果有很大影响,甚至导致假阳性;样品DNA含量在25~40 pg时,所得信号水平显著提高;当样品浓度更高时,超过背景水平,通常会低估检测量。这个联合研究表明,杂交法检测结果与真实的宿主细胞残留DNA含量有很大差异性,并且该方法不稳定,检测时间相对较长[19]。
4.2 荧光染色法
选择只能染色双链DNA的特异性荧光染料,染料与双链DNA结合成复合物,在一定的DNA浓度范围内,荧光强度与DNA浓度成正比,在480 nm波长激发下产生荧光信号,用荧光酶标仪在波长520 nm处进行检测,根据荧光信号强度,计算供试品中的DNA残留量。该方法也是一种DNA总量检测方法,荧光信号易受干扰,特异性较差,因此必须避免环境DNA污染,且所有使用的材料和试剂都必须不含DNA[20]。
4.3 阈值法
阈值法基于两种DNA序列非特异性蛋白即单链DNA(single-stranded DNA,ssDNA)结合蛋白和抗ssDNA的单抗与变性DNA结合,定量检测ssDNA来计算样品中DNA的总量。被生物素标记的结合蛋白与样品中变性的ssDNA结合,同时尿素酶标记的抗ssDNA单抗也可以与ssDNA结合,最终形成的复合物可以被生物素化膜捕获。将膜放入含有尿素溶液的读数仪中,溶液pH值会发生变化,读数仪根据pH值变化计算样品中外源DNA含量。该方法仅限于检测小于800 bp的DNA片段,可能被高浓度DNA(1 ng/ml)抑制,还易受短的DNA片段(20~80 bp)影响,且缺乏稳定性。
4.4 定量PCR法
对外源DNA设计特异性引物和带有荧光标记的探针,或在反应体系中加入灵敏的荧光染料,通过持续监测反应体系中荧光数值的变化,可实时反映特异性扩增产物量的变化。在反应过程中所释放的荧光强度达到预设的阈值时,体系的PCR循环数与该体系所含的起始DNA模板量的对数值呈线性关系。采用已知浓度的DNA标准品,依据以上关系,构建标准曲线,对特定模板进行定量分析,可测定供试品中的外源DNA残留量。有研究比较了SYBR Green荧光染色定量PCR和TaqMan荧光探针定量PCR检测CHO细胞残留DNA的结果,两者检测限度分别为100 fg和10 fg,引物反应效率分别为94.3%〔决定系数(R2)=0.998〕和96.6%(R2=1),结果表明TaqMan探针比SYBR Green染料有更好的灵敏度,该方法可直接用于最终产品中定量检测残留DNA,而无需事先提取DNA[20]。
此外,靶基因组区域的选择对检测结果也有影响。Wang等[10]研究发现选择宿主基因组中多拷贝的靶DNA序列可以增加残留DNA的检出率并准确定量。Liu等[21]针对Vero细胞α-卫星序列,建立了一种可在最低检测限下对几种病毒疫苗进行残留DNA检测的特异性定量PCR法,该方法能够准确显示残留DNA量,确保产品的安全性,同样也可用于记录病毒纯化或灭活过程。
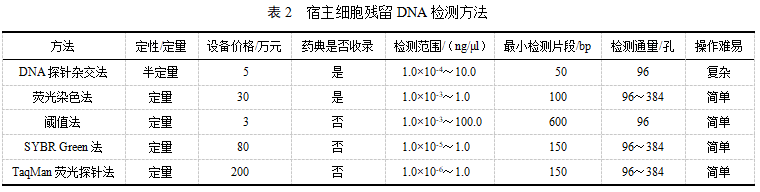
综合以上各方法的原理及用于生物技术药物的科研实例[22-25],得到检测残留DNA的各方法特征及优缺点的比较,见表2。
5
总结与展望
中国药典2015年版收录的宿主细胞残留DNA检测方法只有DNA探针杂交法和荧光染色法,但这两种方法存在灵敏度不高、准确性较差的缺点,已渐渐不能满足产品生产与质量控制的要求。2019年国家药典委员会制定定量PCR标准,作为通则3407外源性DNA残留量测定的第3种方法[26]。这将有利于新药注册申报、工艺优化、质量提高,有利于生物制品安全性得到保证。
一些研究表明,宿主细胞残留DNA除了含量方面,DNA片段大小也是很重要的风险因素。一般认为小于200 bp的DNA片段无致瘤性[27]。研究者们开发了一种基于毛细管凝胶电泳与敏感激光诱导荧光检测残留DNA分子大小的方法,可以检测生物制品中残留DNA的大小,实验表明,大多数宿主细胞残留DNA片段大小为50~2 000 bp。他们从注射方法选择、核酸染料选择、凝胶内染料浓度等方面对该方法进行优化,建立了一种高灵敏性和高准确度的方法,验证表明该方法可用于连续细胞系衍生生物制品生产过程的中间品或原液质量控制。研究者还分析了MDCK细胞生产的流感疫苗样品和Vero细胞生产的呼吸道合胞病毒疫苗样品,证明了上述方法的有效性[28]。这是一种操作简便、数据分析容易、结果明了、具有广泛应用前景的方法。如果该方法可以被国家有关部门或者各生物制药企业广泛使用,将为生物制品残留DNA检测添砖加瓦,使其质量控制更加严格有序。
生物制品的安全性和有效性是评判产品的2个重要指标,近年来,随着人们对健康的关注度越来越高,其安全性尤其备受关注。宿主细胞残留DNA由于其存在致癌性和感染性等风险,需要国家相关部门制定严格的标准,需要质量监控机构建立合理的方法,需要生产厂家建立合理的生产工艺,将潜在危险控制在最低水平,共同助力生物制品研发和生产,为人类造福。
参考文献略
闫璐瑶1,2综述 张家友1,2 杨晓明2,3审校
1武汉生物制品研究所有限责任公司病毒性疫苗研究二室 430207;2国家联合疫苗工程技术研究中心,武汉 430207;3中国生物技术股份有限公司,北京 100029
通信作者:杨晓明,Email: yangxiaoming@sinopharm.com

识别微信二维码,添加生物制品圈小编,符合条件者即可加入生物制品微信群!
请注明:姓名+研究方向!


本公众号所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系(cbplib@163.com),我们将立即进行删除处理。所有文章仅代表作者观点,不代表本站立场。







