

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!
新的机遇和挑战,对研发型生物医药企业药政法规科学工作人士提出了更高的要求。
mao.donglei@PharmaDJ.com
现下国内企业授权转让交易增多,如何跨境生产、分段生产问题突出;MAH落地以来仍存在哪些挑战?如何提升持有人管理和履行契约的能力?法规制定如何集思广益做到可预见性和可持续性?中国药品注册人如何在“内卷”环境下争当一名“斜杠”青年?
上述问题在泰格医药和研发客联合主办的2023年药政法规科学年会上得到了充分讨论。
该会开幕同一天,适逢国家药监局药审中心出台《药审中心加快创新药上市许可申请审评工作规范(试行)》,推高了会场讨论的热情。中国的药物创新如今走上了快车道,而监管环境及政策的变革和优化,带来了新的机遇和挑战,也给研发型生物医药企业药政法规科学工作人士提出了更高的要求。
“为了支持同事们分享和讨论新政策新问题和新策略,增加沟通交流,提升自身能力,泰格医药于2019年发起成立政策法规沙龙,并与研发客合作。”泰格医药政策法规事务副总裁常建青女士说道。

泰格医药政策法规事务副总裁 常建青女士
沙龙发起于2019年,专注于讨论新药临床申报和注册申报相关的政策话题。
拓展阅读
“2021药政法规报告”系列回顾
研发客出版人、总裁戴佳凌在致辞时说:“这些法规背后的思考逻辑是什么,来龙去脉是什么,在落地实施时遇到哪些挑战,还有哪些需要完善之处,研发客总会及时邀请泰格医药政策法规沙龙的资深专家们深度解读。”

研发客出版人、总裁 戴佳凌先生
药渡经纬信息科技有限公司董事长李靖博士用一组数据总结了中国过去创新药研发所取得的成绩。从全球NME/BLA批准上市数量看,中国增速显著高于其他国家/区域,中国新药地位提升。
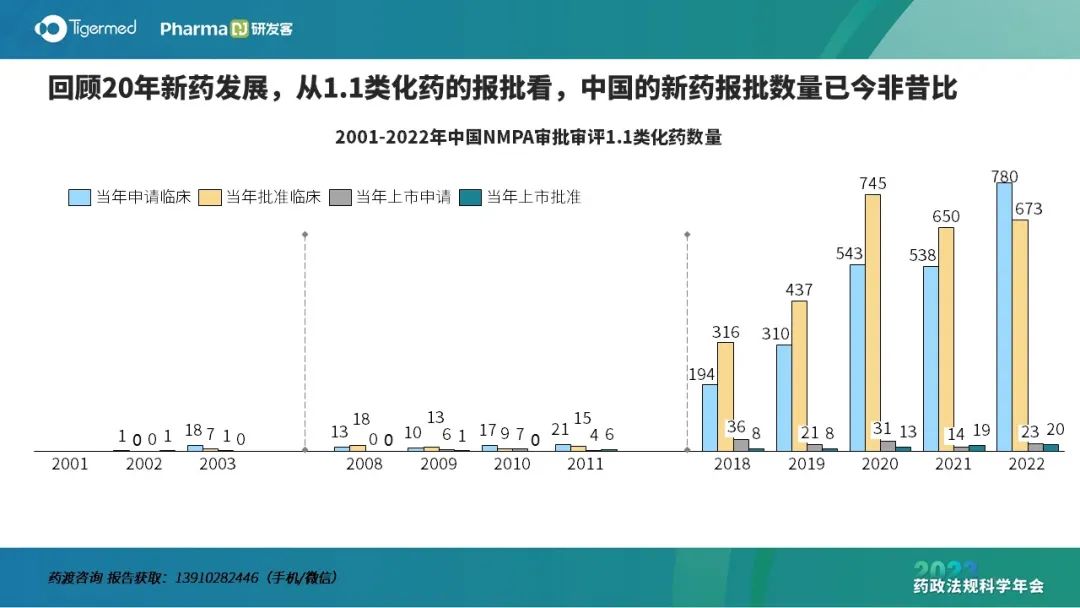
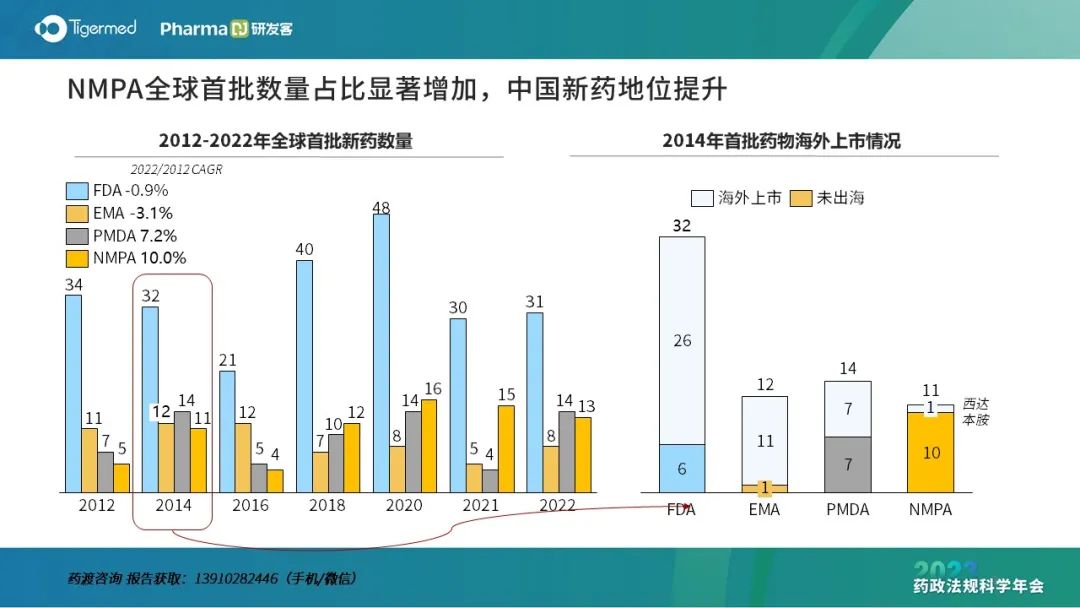
他认为,中国新药近些年取得了巨大成就,但研发质量仍有较大的上升空间,仿创类药物还是主流。2022年中国GDP突破121万亿元,中国医药产业在健康消费梯次升级与疾病谱变迁的共同驱动下具有极高的发展潜力和发展前景。中国医药产业的未来是仿制、仿创与原创的有机结合,根据企业自身资源的差异,布局有所侧重。越“年轻”的企业越应布局原创药物,越“成熟”的企业越应注重仿创、仿制的药物占比。

药渡经纬信息科技有限公司董事长 李靖
“原创药物是医药产业的未来。但仅依靠传统制药技术难以实现从仿到创的转变。靶点选择与化合物的筛选需基于大数据的分析,药渡数据为行业提供全方位的数据支持与数据解读,将与医药企业共同高效完成由仿制迈向创新的产业超越。”李靖说。
MAH落地与药法条例修订进展
2022年5月9日,国家药监局发布《药品管理法实施条例(修订草案征求意见稿)》,对药品研制、生产、经营、使用和监督管理活动的科学要求和伦理准则出台了详细规定。清华大学的杨悦教授介绍了该实施条例的修订历史和最新变化。

清华大学 杨悦教授
值得关注的是2022年5月9日第二次重大修订,已被纳入市场总局立法计划。她建议所有法规制定者都要广泛听取行业意见,不能固化思维,“新的行政许可、制度、程序、措施、义务及法律责任应当不断落实、补充和完善”,同时能清晰向业内传递法规制定的逻辑和理念。
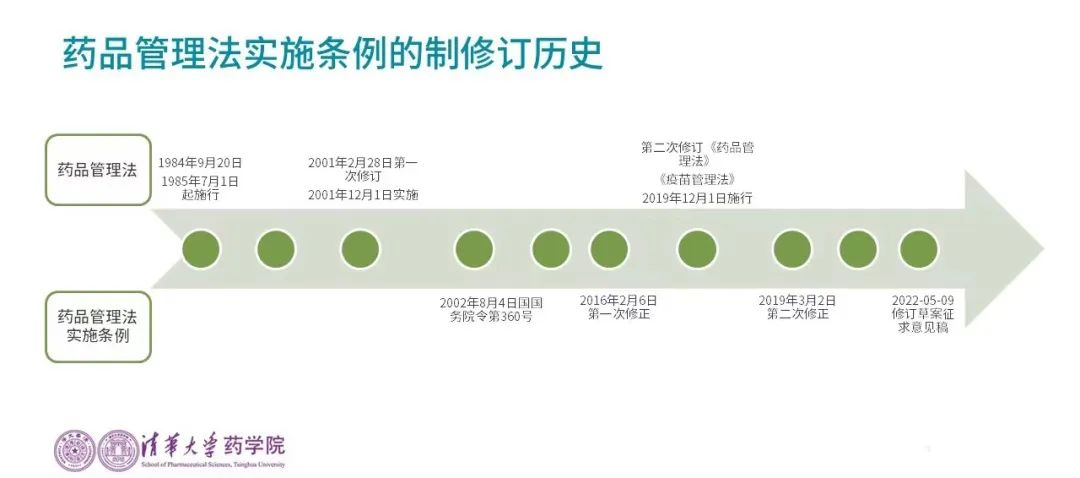
她围绕大家关注的鼓励儿童药、罕见病药物创新的后续政策配套、申请人要具备三大能力、药品再注册、上市许可转让、委托生产中MAH的义务等方面进行了阐述。

现下,国内企业授权转让交易增多,如何跨境生产、分段生产问题突出。杨悦认为,分段生产是解决影响高研发投入药品能否快速产业化,影响国际合作模式的重要话题。与会企业也认为,国家的监管对企业战略制定带来重大影响,应强调对药品上市许可持有人的风险管理,而不是一揽子对原研转让企业一并监管到底。
最后,杨悦强调法规制定需以始为终。“我们制定政策的目的是什么?要达到什么效果?需有法治思维、分类思维、路径思维、程序思维和科学逻辑思维。避免规章条例的冗长与重叠,简化、优化,科学、高效的监管。”杨悦说。
在创新药研发生产过程中,上市许可持有人(MAH)制度激发了创新型药企的研发动力,改善了产能过剩的情况。早在2009年,中国药科大学邵蓉教授团队已致力于中国MAH研究。

中国药科大学 邵蓉教授
“什么是MAH?我国的医药行业现状、推行国际通行的MAH制度的可行性和意义是什么?如果推行MAH,会产生哪些影响?在各环节新增哪些风险,以及如何防控这些风险?当时,国内就此开展的系统性研究是一片空白。”她说。
虽然研究历经“质疑”,但欣喜的是,MAH制度终于在2015年11月在全国十个省(市)试点,并在2019年12月1日实施的新修《药品管理法》中作为核心制度贯穿全法始终。
邵蓉提出,MAH制度的实施在中国确实起到鼓励创新的作用,但MAH制度的精髓却是资源优化与合理配置。“然而,资源优化配置离不开各主体分工协作,这是建立在契约基础上,契约精神与契约能力的保障是MAH制度落地实施的重要前提。在契约精神和契约能力尚待提升完善的眼下,行政这只手还需要发挥重要的调控作用;在契约诚信具备和契约能力提升后,要尊重市场的力量,使得MAH制度政策红利充分发挥出来。”
“MAH实施的挑战体现在:一是‘能力不足’,即表现在持有人及各合作方的契约能力弱,也反映在监管者的监管能力应该加强和提升上;二是全社会对药品风险认知缺乏科学和应有的包容度;三是尚未建立科学的问责机制;四是监管理念有待转变与革新。”邵蓉还强调,持有人的全生命周期的质量安全主体责任要落实到位,药监部门应重点培育和监督持有人的质量管理体系的建设情况,不断提升持有人管理和履行契约的能力。
瓴路药业高级副总裁、注册事务负责人李洁主持了讨论环节。与会者认为,经过过去十多年的药监改革创新,两部大法已出台实施,鼓励创新、科学监管的大方向不会动摇,但不意味着法律一成不变,监管机制仍待建立,监管水平有待提升,监管思维仍待完善,绝不能固化。

瓴路药业高级副总裁、注册事务负责人李洁(左一)、中国药科大学邵蓉教授(左二)、清华大学杨悦教授(左三)、药渡经纬信息科技有限公司董事长李靖
一些基本矛盾,如新药定义、持有人和生产场地跨境、境内外合作模式的问题、分段生产、独占期和数据保护期、解决创仿平衡、解决一窝蜂扎堆研发的问题,仍需要监管和行业不断协商从科学角度解决。共识是,政策制定要有系统性、可预见性和可持续性,指导原则需要优化和完善。“我们期待具有国际视野和战略思维的监管领导者。药监改革正在改革路上,仍需努力并充满信心,携手迈向创新发展。”
争当科学型药品注册人
中国药监局加入ICH以后,标准逐渐国际化。同时,本土企业纷纷走向美国,展开与跨国公司的授权转让与合作。

荣昌生物副总裁 程龙博士
“我们需要全球化和国际化的注册人才。”荣昌生物副总裁程龙表示,企业在海外临床试验和注册事务人才有较大缺口,注册人员需要及早参与研发过程,从临床前研发到Pre-IND会议、IND、Ⅰ期、Ⅱ期、Ⅲ期和上市后研究,制定全生命周期的注册策略规划,提供支持、帮助决策。国际化注册事务一定是全程深度参与,进行相应的全球多中心临床开发。
作为注册人员相关话题讨论的主持人,元羿生物执行副总裁左珺女士认为,在中国药物创新全球化的趋势下,积极参与到药品研发的全过程是注册人员的价值体现。

元羿生物执行副总裁 左珺女士
“新药研发是一个复杂而漫长的过程,是对Biotech研发团队的考验。注册人员需要以法规为基础、以科学为核心帮助和引导研发团队理解监管对以临床价值为导向的药物研发理念,通过创新的策略,共同完成研发的里程碑。”左珺说。
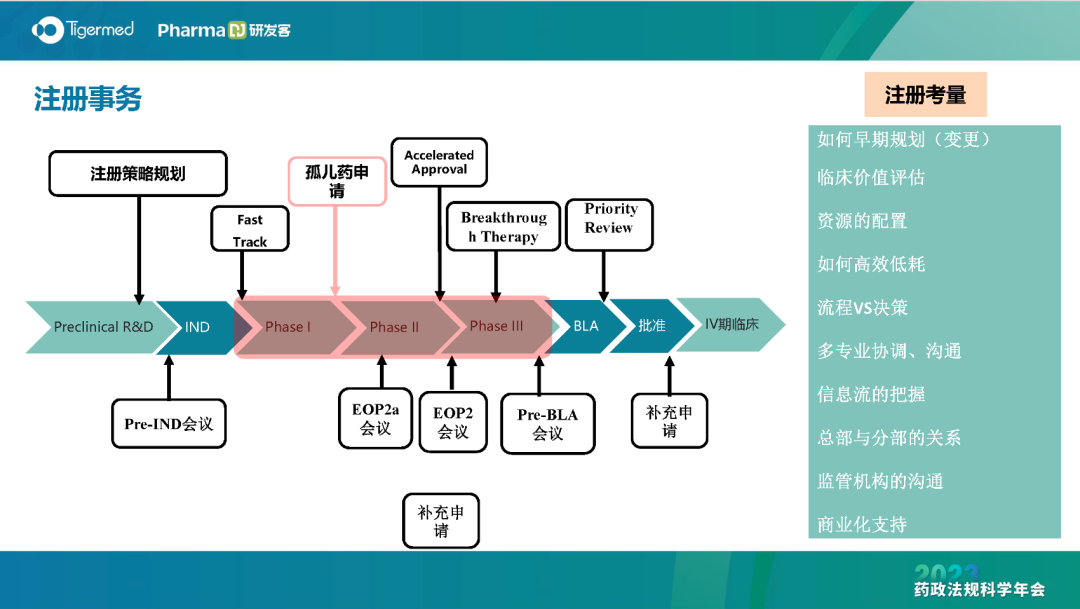
在注册人员的人才发展和职业规划上,程龙介绍说,注册人员是跨学科、跨专业的领域。因此,需要π型人才。所谓π型人才,必须在2个领域深耕:要有广泛的知识面和知识结构,同时有深入的技术技能和精准娴熟的项目经验。
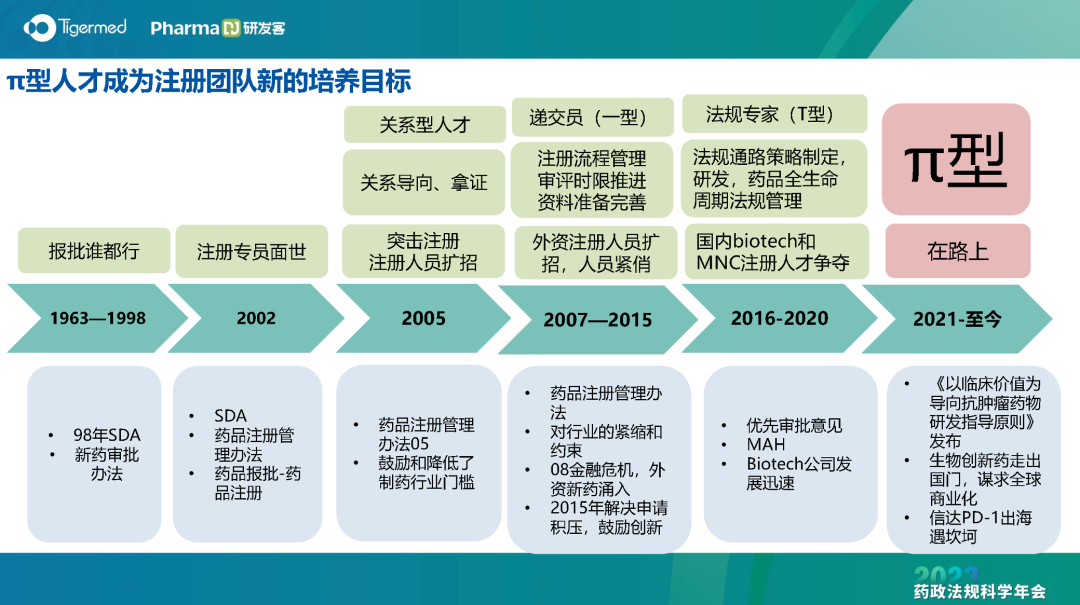
“π型”人才已成为注册团队新的培养目标,需要不断升级打怪。注册人需要前期调研、分析竞品、研读法规,尽早获取信息,撰写注册文件,在时限上分秒必争。
程龙形象地说,注册人既能拿绣花针,也能扛钢枪。同时还是千里眼和顺风耳。由于众望所归,因此注册人无处退缩。“要有唐僧的意志力,孙悟空的能力,猪八戒的人缘以及沙和尚的脚踏实地。”他这样勉励注册人员。
“创新药内卷的情况下,注册人员如何做好斜杠青年?”——再鼎医药高级副总裁蒋燕萍女士的报告题目新颖有趣。

再鼎医药高级副总裁 蒋燕萍女士
蒋燕萍解释了“内卷”的内涵,即低水平复杂化,有增长却没发展,内部竞争激烈,没有对外的扩展和创新。“内卷”十分符合近几年我国新药研发的情况,企业大多采用快速跟进,开发出大量Me-too新药,从“卷仿制”到“卷创新”,研发同质化成为横亘在行业和监管的困扰,对创新药行可持续发展提出严峻挑战,体现在临床试验适应症以及前 10 位靶点的雷同上。
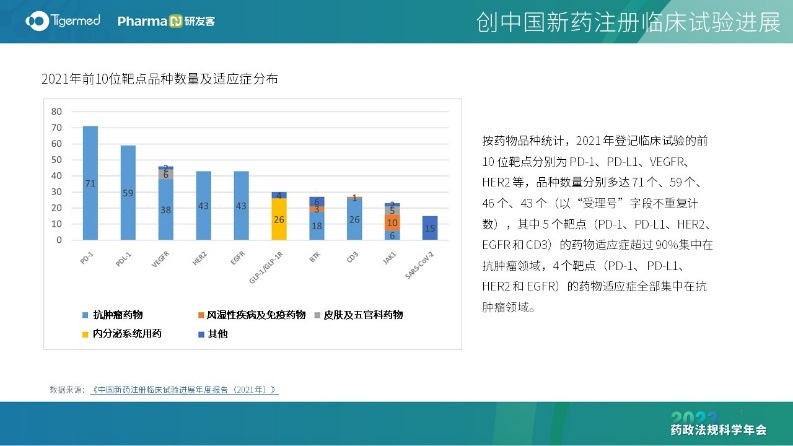
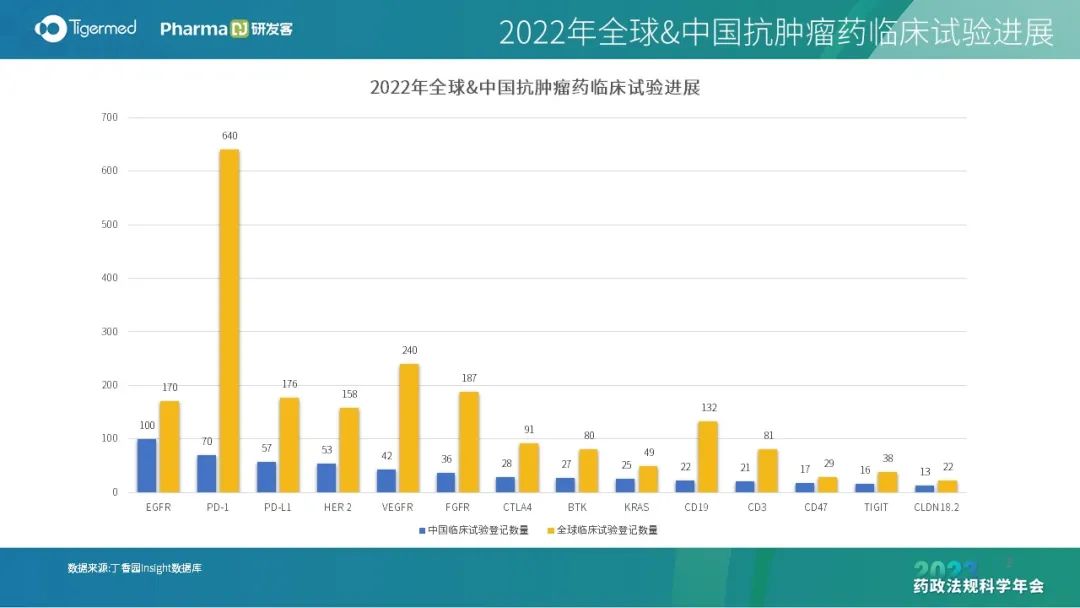
为了破解“内卷”,从基础研究到临床研究都需要变革。例如,CDE发布了《以临床价值为导向的抗肿瘤药物临床研发指导原则》,引导行业回归以患者为中心开发药物;高校源头创新需与产业结合,加强基础研究及转化研究能力,完善高水平人才培养体系,促进科技成果转化。
在这样的前提下,蒋燕萍指出,从仿制到创新,对注册人员的定位和能力要求发生了巨大变化,从过去围绕药学和注册流程的工作到现在更注重临床价值。

“因此,注册人员需要有胜任力,能掌握与解读法规与指南要求,准备、递交新产品的临床研究、上市申请,跟进产品、维护产品生命周期、变更管理和风险评估、管理注册流程和时间表。更深层次的,注册人员要制定产品在中国或全球研发注册战略、参与相关法规和指南制定,为产品开发和公司运营提供各种建议。”
在分享案例环节,蒋燕萍举了一个公司研发的肿瘤生物制品在欧美及亚洲开展临床申报和开发的案例,来阐明注册人员从中国申报到全球申报中在IND策略中起到的关键作用。

她最后建议,注册人员除了拥有注册技术、法规及解读能力、沟通影响力、卓越的执行力还要提升创新能力、增进多学科专业知识、统筹协调和管理能力,卓越执行力。“内卷的情况下,注册人员需提升自己的专业技术、策略、管理能力,争当拥有多重定位的“斜杠青年”,多重多元化的发展能力和价值,拥有怀揣勇闯天下的热忱,做有临床价值的创新药!”蒋燕萍说。

天境生物(杭州)注册事务负责人付洁鹰(左一)主持了圆桌论坛,荣昌生物副总裁程龙(左二)、蒋燕萍(左三)和泰格医药注册部高级总监张小华参加了研讨。
圆桌讨论中,与会嘉宾认为,创新药内卷下,新药研发不能做作业式的研发,要回归研发规律和科学规律,注重质量,从事注册资深同事和年轻同事要积极拥抱变化,不断学习,变成科学家一样的注册人员。以法规为基础,以科学为核心,及早配合药学人员、临床医学人员,从药政法规科学角度为新药研发策略和计划的制定与实施贡献应有的重要价值。
正如瓴路药业的李洁所说,注册人在药物研发中是IND第一棒,NDA最后一棒,责任重大。
加快审评与RWE的真实运用
恒瑞医药注册事务总监朱林介绍了加快审评审批改革进展。

恒瑞医药注册事务总监 朱林博士
近年来,境内加快审批审评经历了四个阶段,包括2015年8月的前开端、2015年8月至2017年10月的解决积压、2017年10月至2020年1月的深化改革,2020年3月至今,突破性治疗药物、附条件批准等加快通道更丰富多样,覆盖IND和上市许可审评期间,但门槛大大提高。
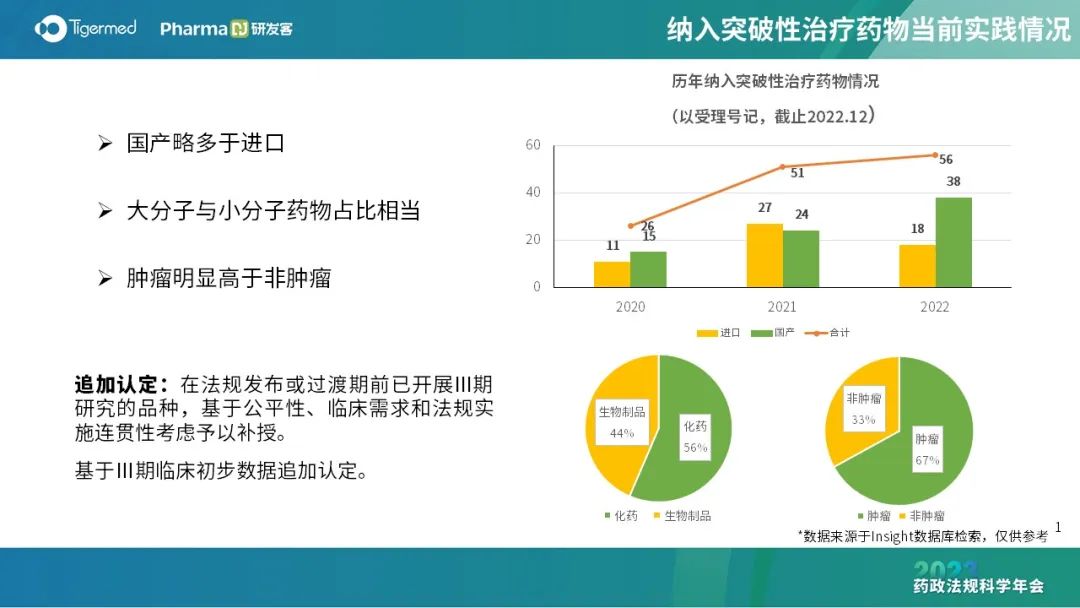
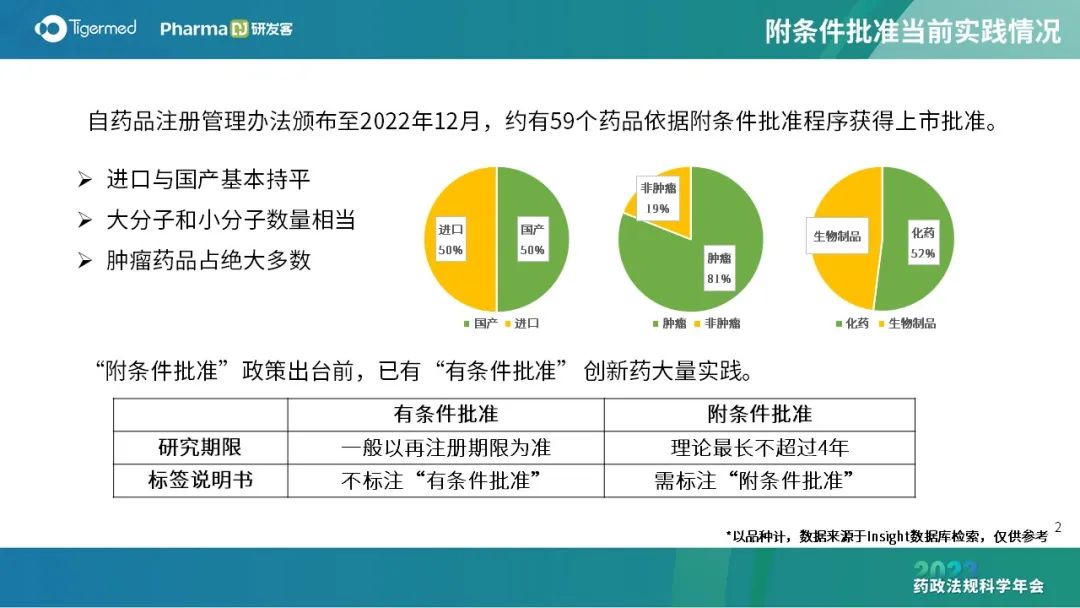
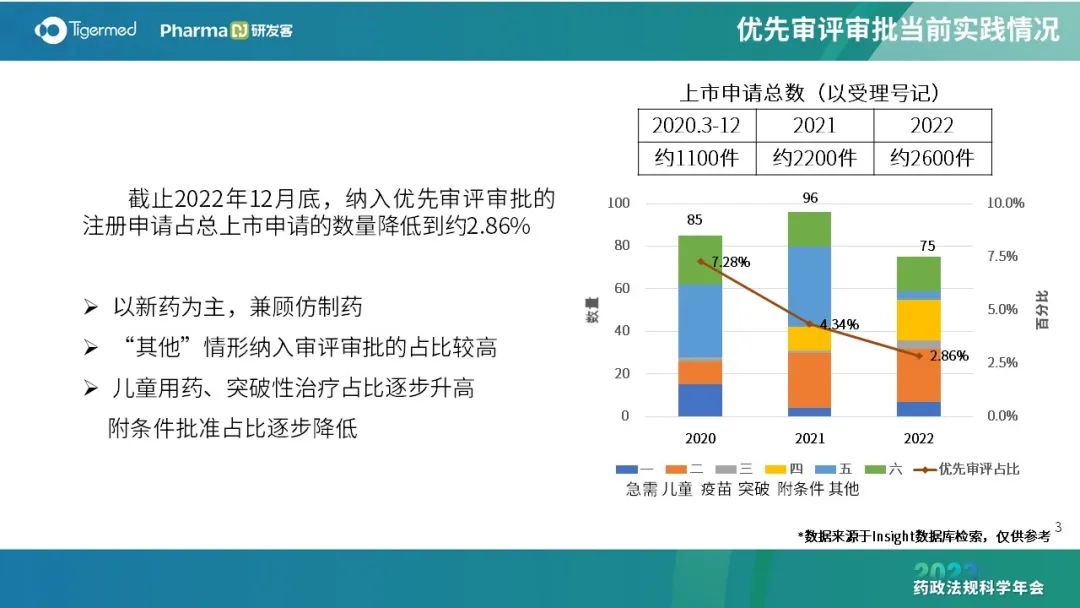
不少创新品种同时被纳入突破、附条件及优先审评,申请人需考虑策略,选择相应适用的加快通道,争取加快获批的政策支持。
“关于突破性治疗药物、附条件批准程序认定的具体意见,期盼CDE能反馈给申请人,以更好理解认定成功或失败的原因,指导后续工作;期待对于纳入突破性治疗药物程序的药物配套更多的激励措施,让这类药物从研发期间就能快起来。”朱林说。(注:会议当天CDE发布的《药审中心加快创新药上市许可申请审评工作规范(试行)》,丰富了对突破性治疗药物资源配置。)
朱林还提出,严重影响生存质量的疾病可否纳入附条件批准,当前附条件批准的范围未包括“严重影响生存质量的疾病”,如系统性红斑狼疮、阿尔兹海默症等也需考虑附条件批准。
“天下武功,唯快不破。新药研发在立项的时候要考虑所有加快程序,加强与CDE沟通交流,将药品更快送到患者手里。”朱林说。
BI亚洲区域流行病学负责人李强博士的报告为《利用真实世界研究支持注册申请》,他介绍了真实世界研究数据、真实世界研究证据的概念和数据来源,如何通过非干预性研究和实效性研究从真实世界数据中得到真实世界证据,传统RCT的优势与不足,例如,由于RCT患者入组需设定入排标准,其代表性受到质疑,如何外推到真实世界就成为挑战等;以及非干预性研究的不足与优势,因此,真实世界研究是RCT一个有力补充,用于注册上市申报。

BI亚洲区域流行病学负责人 李强博士
在药品全生命周期中,真实世界研究/证据贯穿药品开发、成长、成熟阶段。尤其是上市后研究,可以用真实世界研究替代。目前全球50%~60%的真实世界研究来自于上市后研究。
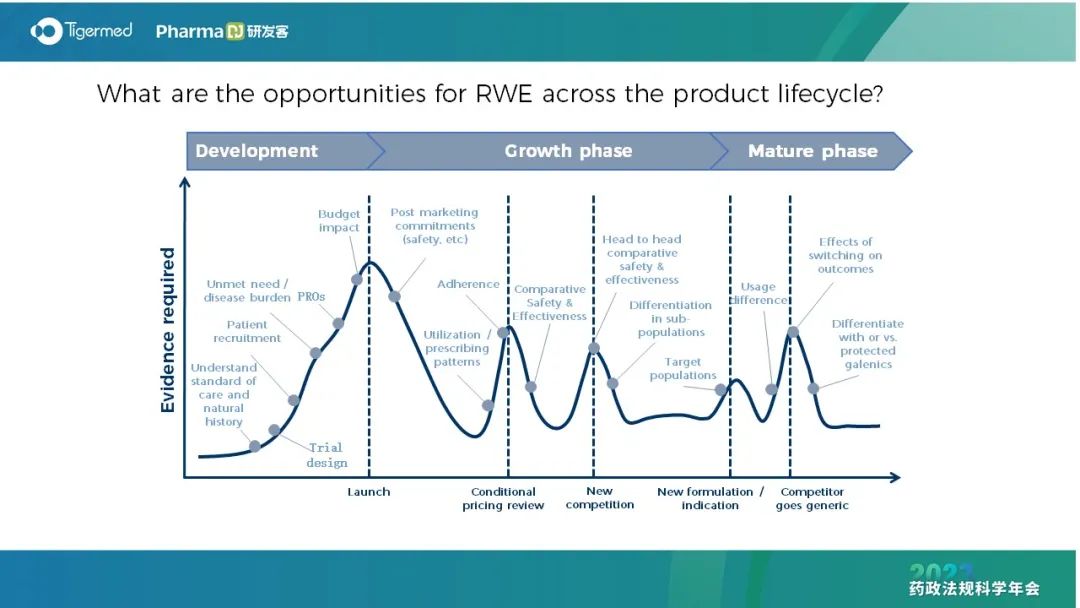
而全球监管机构也越来越意识到真实世界研究对于决策制定时的重要性。
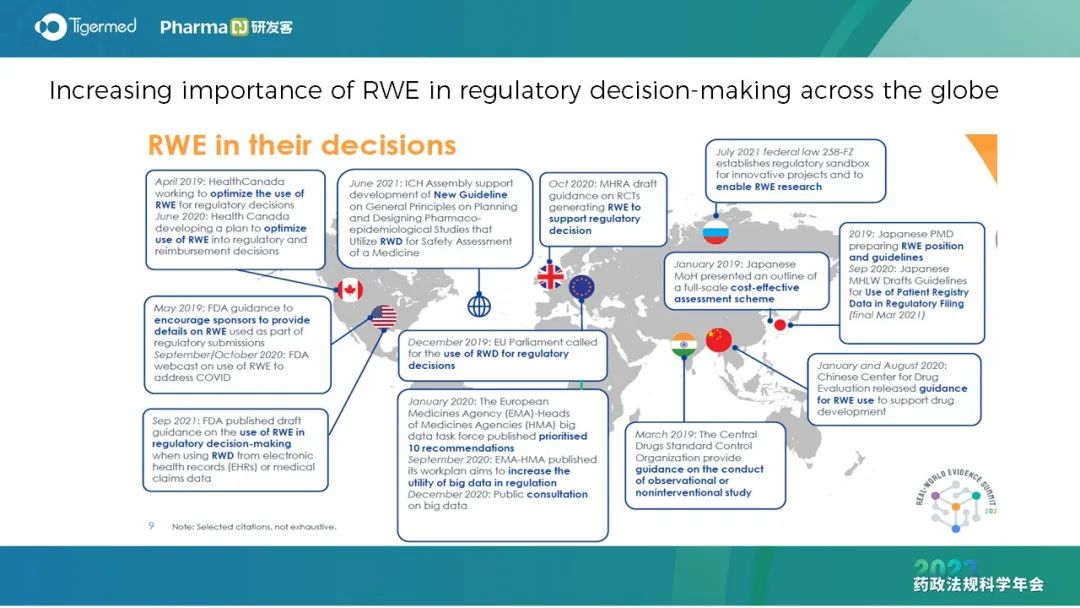
与此同时,中国药监局药审中心也出台了多个真实世界研究相关的技术指导原则。
李强介绍了多个利用RWE数据支持获批的案例。其中,2015年,中国贝伐珠单抗联合卡铂与紫杉醇作为不可切除的晚期、转移性或复发性非鳞状细胞非小细胞肺癌患者的一线治疗。2018年10月,根据3项真实世界研究,贝伐珠单抗被批准通过铂类化疗组合扩大治疗方案。这些研究回顾性分析了来自3家医院的患者数据,结果表明,与单独化疗相比,贝伐珠单抗与铂类化疗联合使用延长了PFS和OS,并且没有发现新的安全问题。该真实世界研究还提供了不同患者亚组分析,如EGFR突变或脑转移患者,从多个角度证实了该药联合治疗的有效性和安全性。
他总结说,基于RWE的扩展需要考虑几个方面的问题:与监管机构开展早期沟通至关重要、数据质量是成功的要素、严谨设计试验方案解决混杂和偏移。

葛兰素史克中国注册事务部负责人邓万和(左一),瑞石生物副总裁、法规事务及临床协调部负责人曹海峰(左二),BI亚洲区域流行病学负责人李强(左三),恒瑞医药注册事务总监朱林
葛兰素史克中国注册事务部负责人邓万和主持了圆桌讨论。与会者讨论了真实世界研究未来的发展方向。目前,国家药监局已在海南博鳌乐城设立了真实世界研究与评价重点实验室,吸引国外企业主动将其产品引入乐城医疗先行区,积累和挖掘数据,作为中国注册审评的有益补充。对于加快审批,曹海峰建议,拟申请附条件批准的创新药沟通交流应尽早开展,并早期规划好药学及临床前研究计划,与可能大大缩短的临床研究时间表相匹配。对获得突破性疗法资格认定的创新药倾斜更多审评资源,沟通交流更精准和细化,从科学层面给予企业具体和全面的指导。


与会嘉宾合影留念。
医药领域的创新,离不开监管政策的规范和指引。泰格医药政策法规沙龙将继续与药政法规科学事务专家学者一起加强交流,分析趋势、探讨问题、分享经验、共赢未来。感谢一百余家本土研发型初创生物医药企业药政法规科学事务负责同事的参与和支持。期待来年再次相聚!
编辑|戴佳凌
dai.jialing@PharmaDJ.com







