

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

各有关单位:
根据国家药品监督管理局2022年度医疗器械注册审查指导原则制修订计划的有关要求,我中心组织编制了《寨卡病毒核酸检测试剂注册审查指导原则(征求意见稿)》(附件1)、《布鲁氏菌IgM/IgG抗体检测试剂注册审查指导原则(征求意见稿)》(附件2)、《丙型肝炎病毒抗体检测试剂注册审查指导原则(征求意见稿)》(附件3)、《基于高通量测序技术的非小细胞肺癌相关基因变异检测试剂临床试验注册审查指导原则(征求意见稿)》(附件4)。现向社会公开征求意见。
如有意见和建议,请填写意见反馈表(附件5),以电子邮件的形式于2022年10月28日前反馈至我中心相应联系人。邮件主题及文件名称请以“《XX注册审查指导原则(征求意见稿)》意见反馈+反馈单位名称”格式命名。
1.寨卡病毒核酸检测试剂注册审查指导原则(征求意见稿)
联系人:程曦
电话:010-86452580
电子邮箱:chengxi@cmde.org.cn
2.布鲁氏菌IgM/IgG抗体检测试剂注册审查指导原则(征求意见稿)
联系人:程曦
电话:010-86452580
电子邮箱:chengxi@cmde.org.cn
3.丙型肝炎病毒抗体检测试剂注册审查指导原则(征求意见稿)
联系人:解怡
电话:010- 86452862
电子邮箱:xieyi@cmde.org.cn
4.基于高通量测序技术的非小细胞肺癌相关基因变异检测试剂临床试验注册审查指导原则(征求意见稿)
本指导原则旨在指导注册申请人对基于高通量测序技术的非小细胞肺癌相关基因变异检测试剂临床试验的设计及开展,同时也为技术审评部门对注册申报资料的技术审评提供参考。
本指导原则是针对基于高通量测序技术的非小细胞肺癌相关基因变异检测试剂临床评价的一般要求,申请人应依据产品的具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。
本指导原则是对申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但需要详细阐明理由,并对其科学合理性进行验证,提供详细的研究资料和验证资料,相关人员应在遵循相关法规的前提下使用本指导原则。
一、适用范围
二、临床试验设计
1.临床试验机构及人员
2.临床试验适用人群和样本类型
适用人群为经病理诊断为NSCLC的人群。应注意纳入NSCLC不同分期的病例,应包括Ⅰ~Ⅳ;同时应纳入不同的组织分型(包括腺癌、鳞癌、腺鳞癌以及大细胞癌)等。阳性样本应包括不同突变率的样本。所纳入人群应在各年龄段均有分布。
3.对比方法的选择
4.最低样本量
临床试验样本量应采用适当的统计学方法进行估算,并详细描述所使用统计方法及各参数的确定依据。此部分临床试验目的为评估两种检测方法之间的一致性,因此建议采用单组目标值法进行最低样本量的估算。通过阳性符合率和阴性符合率来分别计算所需阳性样本和阴性样本的例数,应针对每个基因分别进行样本量的估算。
阴、阳性符合率的临床可接受标准(P0)建议不低于90%。当评价指标PT接近100%时,该样本量估算方法可能不适用,应考虑选择更加适宜的方法进行样本量估算和统计学分析,如精确概率法等。
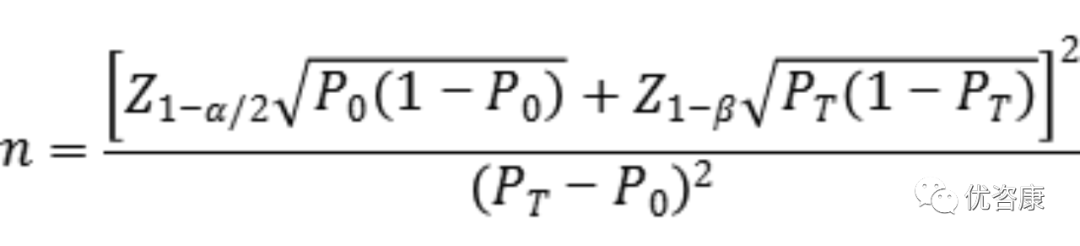
公式中,n为样本量;Z1-α/2、Z1-β为显著性水平和把握度的标准正态分布的分数位,P0为评价指标的临床可接受标准,PT为申报产品评价指标预期值。
应对申报产品所声称的每个基因的各种突变均进行验证,且每种突变类型均应具有一定例数。对于个别临床发生率较低的突变,可结合申报产品的检测原理(如是否共用相同的引物及捕获探针等)及申报产品的检测性能等综合考虑。
5.统计分析
首先应针对所入组人群进行人口学分析,包括性别、年龄、治疗状态、组织分型、分期、入组类型、样本类型(穿刺标本或手术标本)、肿瘤细胞含量、覆盖所有基因亚组等方面进行分析,还应分析临床试验中所纳入的阳性判断值附近的样本情况。
以四格表分别总结申报产品与对比方法的定性检测结果,计算阳性符合率、阴性符合率及相应的95%置信区间。除总体统计外,还要针对每个基因、每种突变进行统计分析,以验证申报产品与对比方法检测结果的一致性。
对于融合,应分别统计每种融合类型的例数,常见的融合类型应有一定例数。
同时应针对存在混合突变的样本单独进行统计分析。
针对不一致样本,应采用合理的方法进行确认,对不一致的原因综合进行分析。
1.具有明确伴随诊断意义的基因变异
针对具有明确伴随诊断意义的基因变异,如EGFR(19del、L858R、T790M)、ALK融合、ROS1融合、MET 14外显子跳跃突变、RET融合等,应参考伴随诊断相关指导原则提供伴随诊断临床证据。
1.1 一致性比对
针对EGFR、ALK融合、ROS1融合,如申报产品为针对已上市的抗肿瘤药物所开发的非原研伴随诊断试剂,则可按照相关指导原则的要求,采用与原研伴随诊断试剂进行一致性比对的方式开展临床试验,也可采用指导原则中的其他评价路径。
1.1.1临床试验机构的数量
针对此部分临床研究,申请人应选择不少于3家经医疗器械临床试验机构备案的临床机构开展临床试验。
1.1.2对比方法的选择
应选用申报产品所声称伴随的抗肿瘤药物的原研伴随诊断试剂作为对比试剂。常见的NSCLC抗肿瘤药物及其所对应的原研伴随诊断试剂如下:
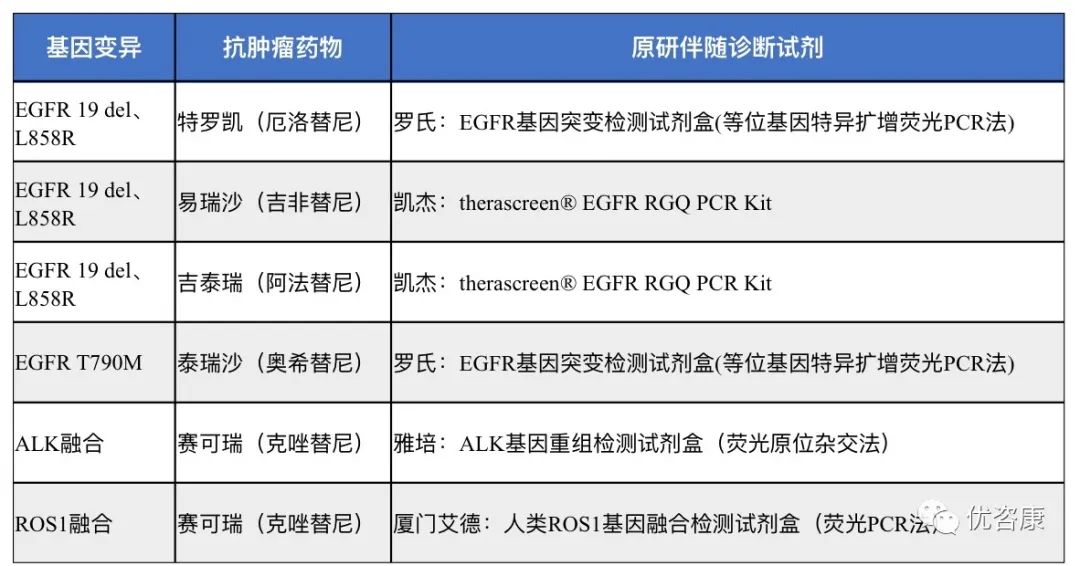
如申报产品为与新开发的抗肿瘤药物同步研发的伴随诊断试剂,则可提供与药物同步开发的药物临床试验资料作为其伴随诊断证据。药物临床试验报告中应明确所使用的伴随诊断试剂为申报产品,并明确申报产品在药物临床试验中所起的作用以及与药物疗效的关系。
针对此类基因变异,对于有已上市同类产品的,无需提供其临床意义的证据。如尚无已上市同类产品的,申请人应提供针对NSCLC患者的临床诊疗指南等临床公认的相关资料作为该类基因变异位点临床意义的支持性资料。
试验方案中应确定严格的病例纳入/排除标准,任何已经入选的病例再被排除出临床试验都应记录在案并明确说明原因。在试验操作过程中和判定试验结果时应采用盲法以保证试验结果的客观性。
委托第三方机构进行参考方法检测的,应提供临床试验机构与第三方机构的委托协议。同时应提供参考方法的详细资料,如:方法原理、所需试剂及仪器、参考方法的性能验证、参考方法质控、典型的实验图谱及数据等。上述资料应由临床试验机构签章确认。
三、参考文献
[1] NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Non-Small Cell Lung Cancer, Version 7.2021 — October 29, 2021.
[2]中国临床诊疗学会(CSCO)非小细胞肺癌诊疗指南 2022.
[3]中国临床肿瘤学会非小细胞肺癌专家委员会,二代测序技术在NSCLC中的临床应用中国专家共识(2020版)[J].中国肺癌杂志,2020,23(9):741-761.
[4]中华医学会病理学分会等,非小细胞肺癌分子病理检测临床实践指南(2021版)[J].中华病理学杂志,2021,50(4):323-332.
[5]《抗肿瘤药物的非原研伴随诊断试剂临床试验注册审查指导原则》(国家药品监督管理局2021年第95号)[Z].
四、起草单位








