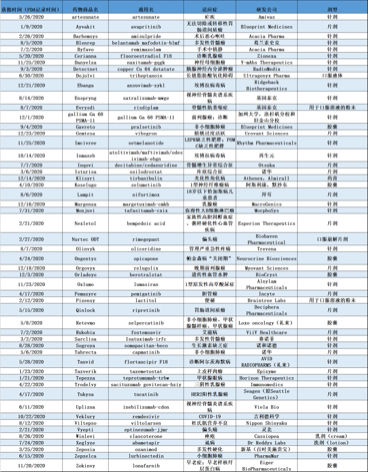
徐佳熹、孙媛媛、黄翰漾、东楠、刘鹭、黄昭宇、杨希成、李博康、王楠、朱新彦、王佳慧、李
昶霖、蔡莹琛(港)、李伟(港)、周逸(港)、余克清(港)
投资要点
•2020年,FDA批准了53种新药,它们分别是新药申请(NDAs)下的新分子实体(NME)或生物制剂许可申请(BLA)下的新治疗生物制剂。这些新药往往是解决未满足医疗需求或显著推进患者治疗的创新产品,它们的活性成分以前从未在美国获得批准。
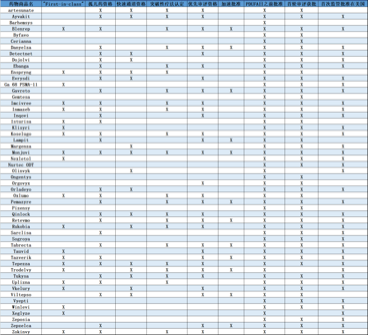
•在53款创新药中,有21款属于“first-in-class”疗法,占总数的40%。这些新药的作用机制不同于已有疗法,有潜力为大众健康带来重要的积极影响,包括新型抗逆转录病毒药物——ViiV Healthcare公司开发的HIV疗法Rukobia(fostemsavir),以及首款用于治疗1型神经纤维瘤病(NF1)的创新疗法Koselugo(selumetinib)等。
•另外,有31款(58%)创新药批准是用于治疗罕见病或孤儿病的(定义为影响少于20万美国患者的疾病)。例如,Evrysdi(risdiplam)是FDA批准的首款治疗脊髓性肌萎缩症的口服疗法。Orladeyo(berotralstat)是预防成人和12岁以上儿科患者的遗传性血管水肿(HAE)发作的首款口服非甾体治疗选择。
|
|
|
|
|
数据来源:FDA,药明康德,兴业证券经济与金融研究院整理
|
在2020年,FDA使用了多种监管通道增强创新药开发和批准的速度和效率。2020年获批的创新药中,32%获得快速通道资格,42%获得突破性疗法认定,57%获得优先审评资格,23%获得加速批准。总体来说,36款创新药(68%)至少获得FDA四大资格认定中的一种。
此外,2020年获批的53款创新药中,92%在第一轮审评过程中获批,75%在美国首先获得监管批准。
|
|
|
|
|
数据来源:FDA,药明康德,兴业证券经济与金融研究院整理
|
Horizon公司IGF-1R靶向单抗Tepezza获批甲状腺眼病适应症
Tepezza最初是由Genmab公司研发的,2017年Horizon Pharma公司获得继续开发该药的权利。2020年1月21日,FDA批准Tepezza®(teprotumumab-trbw)用于治疗甲状腺眼病(Thyroid Eye Disease,TED),这是FDA批准的首个治疗甲状腺眼病的药物,还曾被授予突破性疗法认定、孤儿药资格、快速通道资格、和优先审评资格。此次批准标志着甲状腺疾病治疗的重要里程碑。这种治疗方法有可能改变疾病的进程,通过提供替代性的非手术治疗选择,可能使患者免去需要进行多次侵入性手术的麻烦。
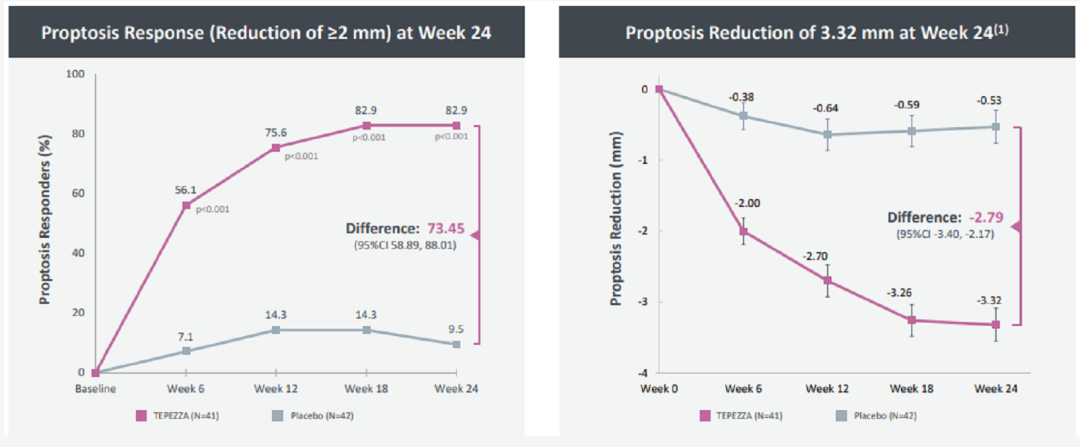
Teprotumumab是一种全人源化胰岛素样生长因子1受体(IGF-1R)单抗,通过与IGF-1R结合并阻断其激活和信号转导,减少炎症并阻止细胞的过度生长。Tepezza是第一种也是唯一一种治疗TED的药物。TED是一种罕见的自身免疫病,人体攻击其自身过表达IGF-1R的眼眶成纤维细胞,使得眼后的肌肉和脂肪组织发炎,导致眼睛往前推并向外凸出。Teprotumumab对于TED的治疗效果非常显著,82.9%的患者眼球直径缩小超过2mm,而对照组仅有9.5%。治疗24周,眼球直径平均缩小3.32mm。2020年10月14日,Horizon Pharma公司新公布了2期临床试验的长期随访数据,表明完成对TED治疗后,疗效持续缓解一年之久。
|
|
|
|
|
数据来源:Science,兴业证券经济与金融研究院整理
|
|
|
|
|
|
|
另外,Teprotumumab也曾进行过治疗乳腺癌、非小细胞肺癌和肉瘤的临床二期研究以及治疗霍奇金、非霍奇金淋巴瘤和实体瘤的临床一期试验,但这些研究目前已终止。
静脉CGRP靶向抗体-灵北Vyepti获批用于成人偏头痛的预防性治疗
2020年4月,灵北(Lundbeck)宣布偏头痛药物Vyepti(eptinezumab)已在美国上市,该药于今年2月21日获得美国FDA批准,用于预防性治疗成人偏头痛。Vyepti通过静脉输注给药,推荐剂量为每季度(3个月)一次100mg,部分患者可能受益于300mg剂量。值得一提的是,Vyepti是第一个也是唯一一个用于预防偏头痛的静脉(IV)疗法,将为患者提供一种有效且普遍耐受的治疗方法,每年仅需静脉输注4次。
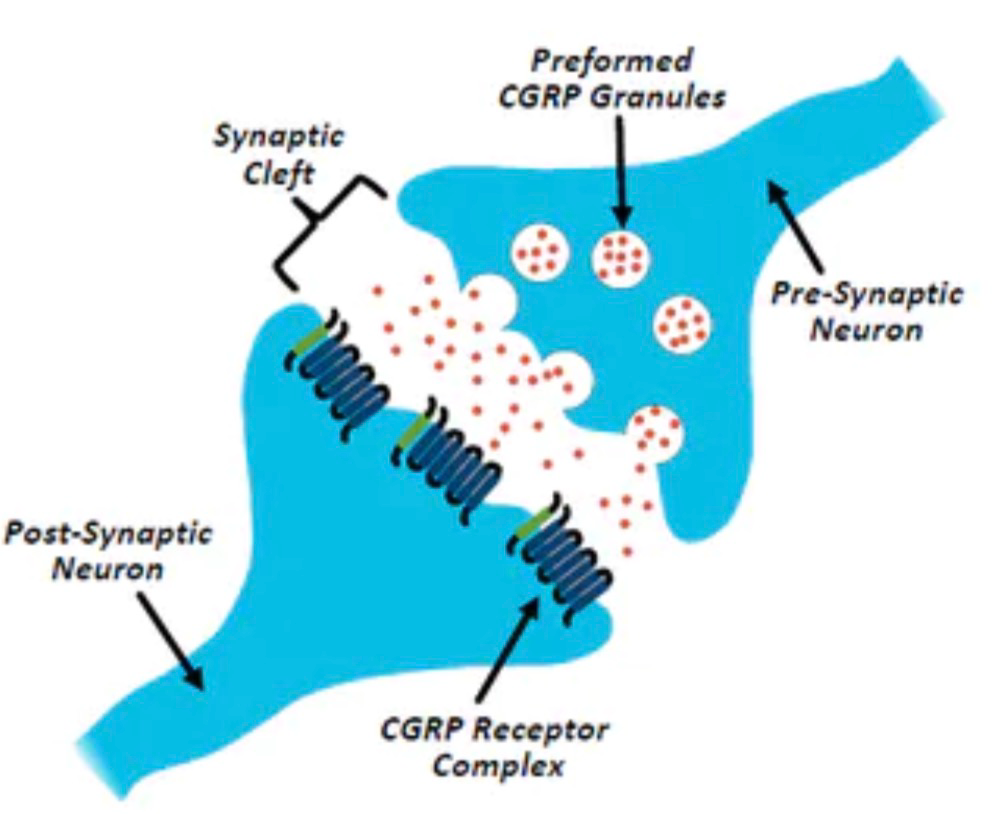
Eptinezumab是Alder公司开发的一种预防偏头痛的单克隆抗体(mAb)。根据公开资料,该产品能够高特异性和强效抑制降钙素基因相关肽(CGRP)。研究显示,CGRP在偏头痛的形成过程中发挥了重要的作用,它可以扩张脑部血管,引起头痛,同时还能参与到痛觉的传递过程中。Eptinezumab通过特异性地结合CGRP,阻止偏头痛的产生和发展。目前,CGRP及其受体已成为偏头痛药物研发的热门靶点。
|
|
|
|
|
数据来源:PubMed,兴业证券经济与金融研究院整理
|
Biohaven公司口服偏头痛新药Nurtec ODT成功获批上市
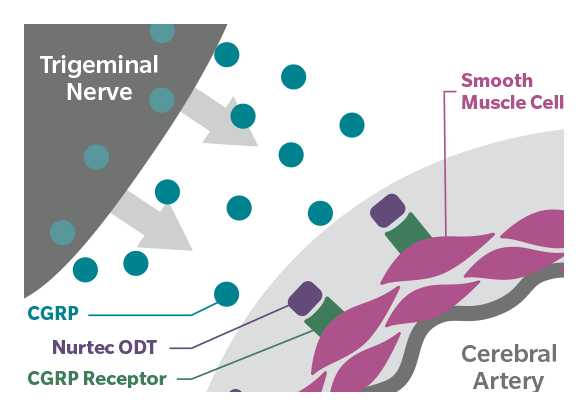
2020年02月27日,FDA批准Biohaven制药公司的Nurtec ODT(rimegepant),用于成人偏头痛(有或无先兆)急性治疗。该药可在口腔内立即分散,而不需要水,可以非常方便的、随时随地服用。Nurtec ODT的上市,将为偏头痛患者提供一种重要的、新的口服急性治疗药物,能迅速减轻和消除疼痛、恢复生活。但Nurtec ODT不适用于偏头痛的预防性治疗。
值得一提的是,Nurtec ODT是首个也是唯一一个获FDA批准的速效口腔崩解片(ODT)剂型的降钙素基因相关肽(CGRP)受体拮抗剂,通过可逆阻断CGRP受体,从而抑制CRP神经肽的生物活性。CGRP及其受体在与偏头痛病理生理学相关的神经系统区域表达。CGRP受体拮抗作用是偏头痛急性治疗的一种新的作用机制,与现有的曲坦类(血清素1B/1D激动剂)和阿片类药物的作用机制明显不同。在临床研究中,单次口服Nurtec ODT 75mg,可在1小时内迅速缓解疼痛,恢复正常功能,对许多患者的持续疗效可达48小时;此外,接受单剂量Nurtec ODT治疗的患者中,高达86%的患者在24小时内没有使用偏头痛抢救药物。
|
|
|
|
|
数据来源:PubMed,兴业证券经济与金融研究院整理
|
多发性骨髓瘤新药赛诺菲CD38抗体Sarclisa获FDA批准
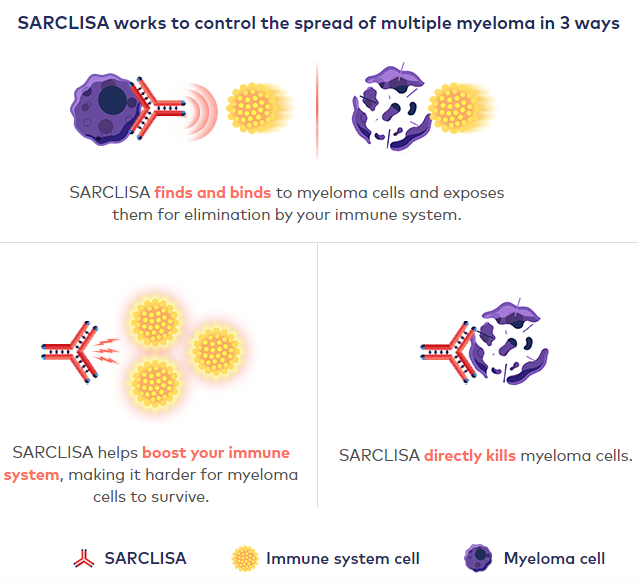
美国食品药品监督管理局于2020年3月2日批准赛诺菲公司新药Sarclisa(isatuximab-irfc)与泊马度胺(pomalidomide)和地塞米松(dexamethasone)联合用药用于成人治疗多发性骨髓瘤(multiple myeloma),适用于之前已经用过至少两种药物(包括来那度胺[lenalidomide]和1种蛋白酶体抑制剂)治疗但无效的患者。随后,Sarclisa于2020年6月初获欧盟委员会(EC)批准。英国国家卫生与临床优化研究所(NICE)已支持赛诺菲Sarclisa (isatuximab)用于四线治疗多发性骨髓瘤(MM)患者。
多发性骨髓瘤(MM)是第二常见的血液癌症,在美国影响了超过13万患者,每年约有3.2万美国人被诊断为多发性骨髓瘤。大多数患者最终不幸复发,对目前可用的疗法难治。Sarclisa联合泊马度胺和地塞米松(pom-dex)方案,将为这些患者提供一个重要的新治疗选择。
Sarclisa剂型为静脉滴注剂,它的活性药物成分isatuximab是一种IgG1嵌合单克隆抗体,靶向浆细胞CD38受体的特定表位,能够触发多种独特的作用机制,包括促进程序性肿瘤细胞死亡(凋亡)和免疫调节活性。CD38在多发性骨髓瘤(MM)细胞上呈高水平表达,是MM和其他恶性肿瘤中抗体治疗的细胞表面受体靶标。在美国和欧盟,isatuximab均被授予了治疗R/R MM的孤儿药资格。目前,赛诺菲也正在评估isatuximab治疗其他血液系统恶性肿瘤和实体瘤的潜力。
FDA批准Immunomedics公司ADC药物Trodelvy用于三阴性乳腺癌治疗
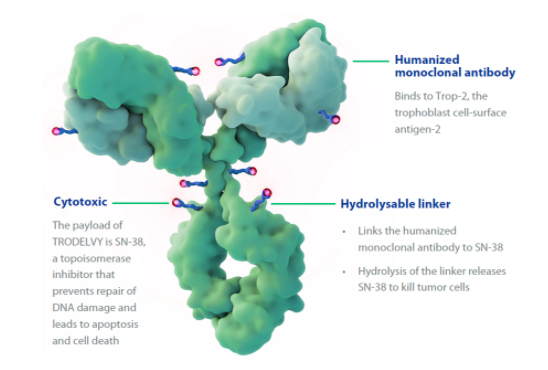
4月22日,Immunomedics公司宣布,美国FDA批准Trodelvy(sacituzumab govitecan-hziy),用于先前已接受过至少两种疗法治疗的转移性三阴性乳腺癌(mTNBC)成人患者。值得一提的是,Trodelvy是FDA批准的第一个专门治疗复发或难治性mTNBC的ADC药物,也是FDA批准的第一个抗Trop-2 ADC药物。之前,FDA授予了Trodelvy突破性药物资格(BTD)和优先审查。FDA批准Trodelvy是基于一项临床试验的结果,该试验纳入108名转移性三阴性乳腺癌患者,这些患者既往至少接受过2种治疗。结果显示,Trodelvy的总缓解率为33.3%,缓解的中位持续时间为7.7个月。在缓解的患者中,55.6%的患者疗效维持6个月或以上,16.7%的患者疗效维持1年及以上。
Trodelvy的活性药物成分为sacituzumab govitecan,是一种新型、首创的抗体药物偶联物(ADC)药物,由靶向TROP-2抗原的人源化IgG1抗体与化疗药物伊立替康(一种拓扑异构酶I抑制剂)的代谢活性产物SN-38偶联而成。TROP-2是一种在90%以上的TNBC中表达的细胞表面糖蛋白,但在正常组织中表达有限。因此Trodelvy可以通过特异性靶向Trop-2单抗Sacituzumab将临床常用化疗药物伊立替康的活性代谢产物govitecan(SN-38)靶向运送到实体瘤病灶,发挥化学毒性杀伤作用。TNBC是一种预后很差的侵袭性癌症,5年生存率不到15%。除了传统化疗之外,治疗方案极其有限。Trodelvy有潜力成为TNBC治疗的一个标准护理药物。目前,Immunomedics公司正在评估Trodelvy治疗多种类型癌症,包括mTNBC、尿路上皮癌、非小细胞肺癌等。
2020年4月21日,中国国家药监局药品审评中心(CDE)最新公示,Everest公司提交的Trodelvy在中国获批一项临床试验,适应症为“接受过至少2线既往治疗的转移性三阴性乳腺癌”。这意味着随着Trodelvy获得FDA的加速批准,在不久地将来中国关于Trodelvy的临床试验将会如陆续地展开。
Viela Bio抗CD19单抗Uplizna获批治疗视神经脊髓炎谱系疾病
2020年06月12日,美国FDA宣布,批准Viela Bio公司的抗CD19单克隆抗体Uplizna(inebilizumab-cdon)上市,治疗视神经脊髓炎谱系疾病(NMOSD)患者,这些患者体内携带靶向AQP4水通道蛋白的抗体。Uplizna是目前第一个也是唯一1个被批准用于治疗抗Aquaporin-4(AQP4)抗体阳性的NMOSD成年患者的B细胞耗竭剂。FDA曾授予inebilizumab治疗NMOSD的孤儿药称号和突破疗法。
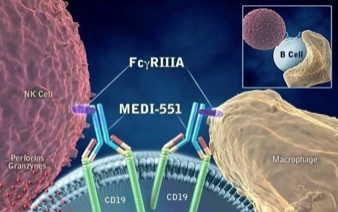
NMOSD是一种罕见的中枢神经系统自身免疫性疾病,其中免疫系统细胞和自身抗体会攻击并损害视神经和脊髓。NMOSD主要与一类自身抗体—AQP4-IgG有关,该类抗体由B细胞分化的浆母细胞和浆细胞产生,其能结合广泛存在于中枢神经系统的星形胶质细胞,由此触发免疫细胞攻击。CD19广泛表达于B细胞表面和其分化形成的浆母细胞/浆细胞表面,较只表达在B细胞表面的CD20更广泛。Inebilizumab靶向CD19,利用Fc部分招募NK细胞和巨噬细胞,通过ADCC和ADCP消耗表达CD19的B细胞(包括浆母细胞和浆细胞),减少抗原呈递并减少促炎症因子。
在230位成年患者的临床研究中证明了Uplizna治疗NMOSD的有效性,该研究评估了静脉Uplizna的疗效和安全性。在该试验中,230名患者中有213名患者具有针对AQP4的抗体(抗AQP4抗体阳性)。在197天的研究中,与安慰剂治疗组相比,接受Uplizna治疗的161名抗AQP4抗体阳性患者的NMOSD复发风险降低了77%。没有证据表明抗AQP4抗体阴性的患者受益。
2019年5月28日,豪森药业与Viela Bio达成了战略合作,在中国开发Inebilizumab治疗NMOSD以及其他潜在的炎症/自身免疫和血液学恶性肿瘤适应症。
Monjuvi联合来那度胺治疗B细胞淋巴瘤获FDA批准
2020年7月31日,MorphoSys和Incyte联合宣布,美国FDA加速批准了Monjuvi(tafasitamab-cxix,MOR208),联合来那度胺(lenalidomide),用于治疗不适合自体干细胞移植(ASCT)的复发或难治性弥漫性大B细胞淋巴瘤(r/r DLBCL)成人患者,包括源于低级别淋巴瘤的DLBCL。这是首个获批用于一线治疗期间或治疗后病情进展的r/r DLBCL成人患者的二线疗法。此前,FDA已授予Monjuvi快速通道、突破性治疗和优先评审的资格。
Monjuvi(tafasitamab-cxix,MOR208)是一种新型人源化Fc结构域优化的溶细胞性CD19靶向性免疫增强单克隆抗体,其Fc结构域进行了修饰(包含2个氨基酸取代S239D和I332E),通过提高对效应细胞上激活型FcγRIIIa的亲和力,显著增强抗体依赖性细胞介导的细胞毒性(ADCC)和抗体依赖性细胞吞噬(ADCP),从而改善肿瘤细胞杀伤的关键机制。临床前模型研究中,tafasitamab已被证实通过结合CD19可诱导癌细胞直接凋亡,CD19是多种B细胞恶性肿瘤的一个明确生物标志物。
目前,tafasitamab正被开发用于2种B细胞恶性肿瘤,包括慢性淋巴细胞白血病(CLL)和DLBCL。在全球范围内,CLL是成人中最常见的白血病类型,DLBCL是成人中最常见类型的非霍奇金淋巴瘤(NHL),占所有NHL病例的40%。DLBCL是一种影响免疫系统B细胞的侵袭性疾病,其特征是淋巴结、脾脏、肝脏、骨髓或其他器官中的恶性B细胞迅速生长。30%-40%的患者对初始治疗无应答或之后复发,对有效的治疗方法存在着显著未满足的医疗需求。在美国,每年约有10000例患者被诊断为不符合ASCT条件的r/r DLBCL。
tafasitamab上市后,将挑战市面上治疗R/R DLBCL的2款抗CD19 CAR-T疗法——诺华Kymriah和吉利德Yescarta。疗效方面,tafasitamab与Kymriah和Yescarta具有可比性。用药方面,Kymriah和Yescarta均需针对每例患者单独制备,需要耗费一定时间,tafasitamab则是一种工业化生产的即用型单抗,随取随用。治疗成本方面,Kymriah和Yescarta均定价数十万美元,而tafasitamab可以控制的非常低。
全球首个BCMA靶向药葛兰素史克骨髓瘤新药BLENREP在美获批
2020年8月5日,美国食品和药物管理局(FDA)批准葛兰素史克(GSK)Blenrep(belantamab mafodotin,GSK2857916)。该药是一种靶向B细胞成熟抗原(BCMA)的抗体药物偶联物(ADC),作为一种单药疗法,用于治疗先前已接受过至少4种疗法(包括一种免疫调节剂、一种蛋白酶体抑制剂、一种抗CD38抗体)的复发或难治性多发性骨髓瘤(MM)患者。Blenrep是全球获批的第一个BCMA靶向疗法,有望改善复发或难治性骨髓瘤患者无药可用的难题。。
belantamab mafodotin是一种新型人源化Fc-改造过的抗BCMA单抗与细胞毒制剂MMAF(monomethyl auristatin-F)通过一种非裂解链接子(药物链接技术从西雅图遗传学取得授权)偶联而成的ADC药物。belantamab mafodotin通过抗BCMA单抗靶向结合MM细胞表面的BCMA,之后迅速被MM细胞内化,在溶酶体中降解并在MM细胞内释放出非渗透性的MMAF发挥作用。MMAF是一种有丝分裂抑制剂,为抗微管蛋白化合物,能通过阻断微管聚合抑制细胞分裂,可使肿瘤细胞停止于G/M期并诱导caspase-3依赖的细胞凋亡。此外,belantamab mafodotin还能诱导NK细胞介导的ADCC(抗体依赖性细胞介导的细胞毒性作用),同时诱导巨噬细胞介导ADCP(抗体依赖性细胞介导的吞噬作用)。
Blenrep的获批,是基于DREAMM-2研究的6个月初步结果,该研究纳入了患有复发性或难治性多发性骨髓瘤的患者,这些患者尽管接受了标准治疗,但病情仍在恶化。在研究中,接受中位既往7线治疗的患者(n=97)中,Blenrep的总缓解率(ORR)为31%(97.5% CI;21-43);6个月分析时尚未达到中位缓解持续时间(DoR),73%的缓解者的DoR等于或大于6个月。
第三款视神经脊髓炎谱系障碍创新药罗氏IL-6R单抗Enspryng获批
2020年08月14日,美国食品和药物管理局(FDA)已批准罗氏(Roche)Enspryng(satralizumab),用于治疗抗水通道蛋白-4(AQP4)抗体阳性的视神经脊髓炎谱系障碍(NMOSD)成人患者。NMOSD是一种罕见的、终生的、使人衰弱的中枢神经系统自身免疫性疾病,常被误诊为多发性硬化症(MM)。NMOSD主要损害视神经和脊髓,导致失明、肌无力和瘫痪。
Enspryng是第一个也是唯一一个获FDA批准治疗AQP4抗体阳性NMOSD的皮下治疗方案,可由患者自己或护理人员,每4周皮下注射一次。同时,Enspryng是第一个也是唯一一个靶向抑制白细胞介素-6受体(IL-6R)活性治疗NMOSD的治疗方案。在2项关键III期研究中,Enspryng作为一种单一疗法和作为基线免疫抑制剂治疗(IST)的附加疗法,在广泛的NMOSD患者群体中显示了强大的疗效,并显著降低了复发的风险。
Enspryng由罗氏旗下中外制药(Chugai Pharma)采用新型抗体回收技术开发。与传统技术相比,这种技术可以延长抗体循环的持续时间,最大限度地抑制IL-6信号,同时将慢性病中的安全风险降至最低。NMOSD患者会经历不可预测的严重复发,直接导致累积的、不可逆的神经损伤和残疾。通过早期治疗预防复发,可对预防残疾产生积极影响,这是NMOSD疾病管理的首要目标。
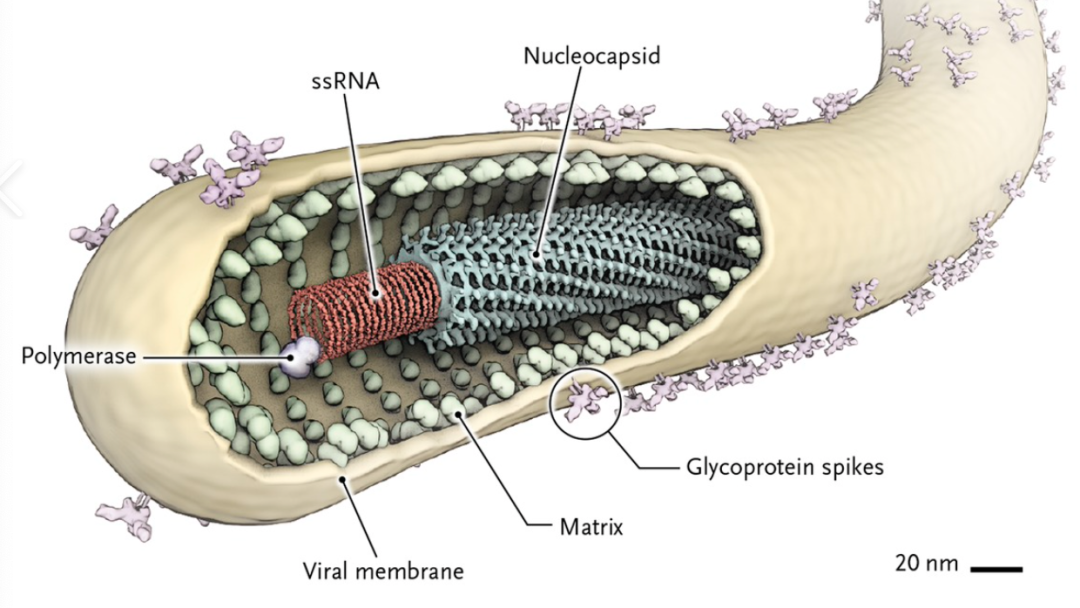
首个治疗埃博拉病毒药物再生元Inmazeb获FDA批准
2020年10月14日,美国食品药品监督管理局(FDA)批准了再生元(Regeneron)公司研发的三种单克隆抗体的混合物抗体鸡尾酒疗法Inmazeb(atoltivimab、maftivimab和odesivimab-ebgn,曾用名REGN-EB3)上市,用于治疗成人及儿童埃博拉病毒感染。这是FDA批准的首款治疗埃博拉病毒感染的药物。Inmazeb已获得“孤儿药”认定,FDA也授予了它突破性疗法的认定。
埃博拉(Ebola virus)是一种十分罕见的病毒,它能引起人类和其他灵长类动物产生埃博拉出血热的烈性传染病病毒,其引起的埃博拉出血热(EBHF)是当今世界上最致命的病毒性出血热,感染者有恶心、呕吐、腹泻、肤色改变、全身酸痛、体内出血、体外出血、发烧等症状。死亡率在50%至90%不等,致死原因主要为中风、心肌梗塞、低血容量休克或多发性器官衰竭。Inmazeb通过靶向埃博拉病毒表面的糖蛋白,阻止其侵入人体内。这种糖蛋白通过与细胞表面的受体相结合,导致病毒和宿主细胞膜的融合,使病毒进入细胞。组成Inmazeb的三种抗体可同时与这种糖蛋白结合,阻断病毒附着和进入细胞。Inmazeb由再生元利用专有的VelociSuite®快速反应技术开发,目前该技术正被应用于开发针对新型冠状病毒肺炎(COVID-19)的新型抗体鸡尾酒疗法。REGN-EB3相比瑞德西韦、mAb114以及ZMapp能明显降低埃博拉病毒感染的死亡率,治疗28天的死忙率为33.5%,显著低于瑞德西韦的53.1%和ZMapp的49.7%。
|
|
|
|
|
数据来源:PubMed,兴业证券经济与金融研究院整理
|
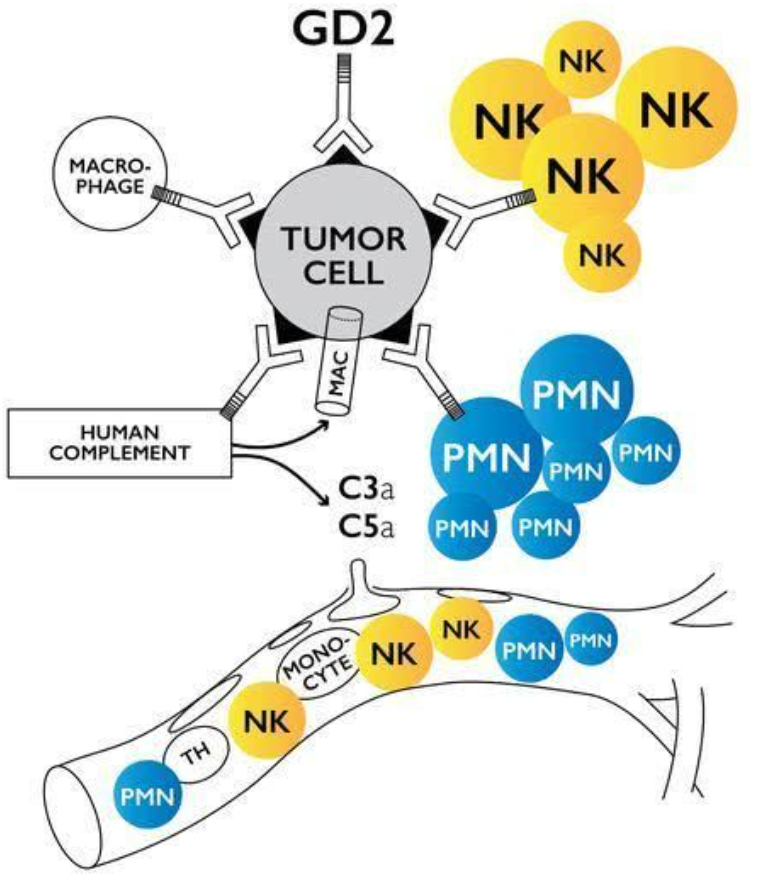
Y-mAbs公司GD2单克隆抗体Danyelza获批用于神经母细胞瘤的治疗
11月25日,Y-mAbs公司开发的GD2单克隆抗体Danyelza (naxitamab-gqgk) 经FDA加速审批条例获批上市,用于与粒细胞-巨噬细胞集落刺激因子(GM-CSF)联合治疗对既往治疗表现出部分缓解、轻微缓解或疾病稳定的复发/难治性高危神经母细胞瘤儿童(1岁及以上)和成人患者。Danyelza曾被FDA授予优先审评资格、孤儿药资格、突破性疗法认定和罕见儿科疾病认定。
Danyelza的批准得到了两项关键性研究的临床试验证据的支持,这两项研究在复发/难治性高危神经母细胞瘤患者中开展。Danyelza耐受性良好,在临床试验中很少中断治疗,不良事件在临床上可管理。Y-mAbs递交naxitamab的上市申请数据显示,该疗法使患者达到78%的客观缓解率(ORR),并使50%的患者无进展生存期(PFS)达到24个月。
神经母细胞瘤是一种极具侵袭性的肿瘤,是婴幼儿最常见的肿瘤之一,尽管近年来强化多模式治疗已提高了生存率,但幸存者有很高的复发风险。DANYELZA是一种靶向神经节苷脂GD2的人源化单克隆抗体,该神经节苷脂GD2在各种神经外胚层衍生的肿瘤和肉瘤中高度表达。GD2抗原在各种神经外胚层来源的肿瘤和肉瘤中呈现高表达,包括神经母细胞瘤、黑色素瘤和骨肉瘤等肿瘤。Danyelza通过与肿瘤表面的GD2抗原结合,能够触发抗体介导的细胞毒性反应并激活免疫系统中的补体系统,从而达到杀伤肿瘤的效果。目前,Danyelza也正被开发用于治疗骨肉瘤以及其他GD2阳性肿瘤。在门诊患者中,DANYELZA一周给药3次,每四周重复一次治疗。
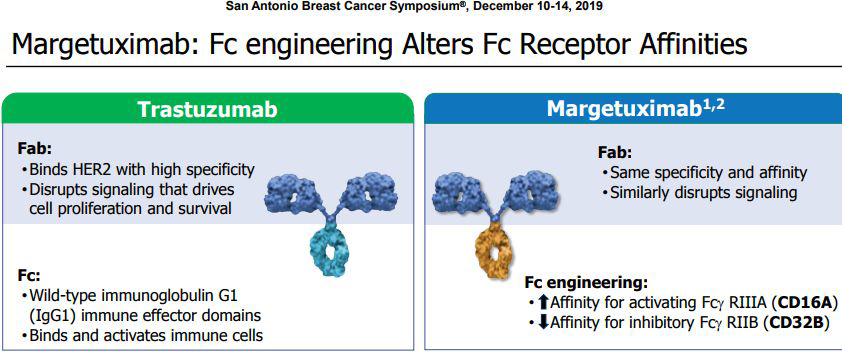
FDA批准MacroGenics公司Margenza联合化疗治疗HER2+转移性乳腺癌
2020年12月16日美国FDA批准免疫优化的抗HER2单克隆抗体Margenza(margetuximab-cmkb)上市,与化疗联合用于转移性HER2阳性乳腺癌成人患者的治疗,这些患者先前接受过两种或两种以上抗HER2方案(其中至少一种为治疗转移性疾病)。Margenza是第一个在头对头3期临床试验中与罗氏王牌生物制剂Herceptin(赫赛汀,通用名:trastuzumab,曲妥珠单抗)相比显著改善无进展生存期(PFS)的HER2靶向疗法,该药的批准上市,将为HER2阳性转移性乳腺癌患者群体带来一种新的治疗选择。
人表皮生长因子受体2(HER2)是在一些癌细胞表面发现的一种促进生长的蛋白,与侵袭性疾病和不良预后有关。大约15-20%的乳腺癌病例为HER2阳性。靶向HER2的单克隆抗体极大地改善了治疗结局,然而,大量患者仍然会出现疾病进展,需要创新疗法。Margenza是MacroGenics公司管线中获批的首个产品,其通过MacroGenics的Fc优化技术进行了工程设计,以增强其免疫系统的参与,导致更强的体外抗体依赖性细胞介导的细胞毒性作用(ADCC)和自然杀伤(NK)细胞活化,提高对癌细胞的杀伤力。
SOPHIA试验结果表明,与曲妥珠单抗联合化疗相比,margetuximab联合化疗可使疾病进展或死亡的风险降低24%,中位PFS为5.8个月,而曲妥珠单抗联合化疗组的中位PFS为4.9个月。margetuximab联合化疗的中位OS为22%,而曲妥珠单抗联合组的中位OS为16%。
FDA批准第二款埃博拉疗法:Ridgeback单抗药物Ebanga
2020年12月21日,美国食品药品监督管理局(FDA)批准了Ridgeback Therapeutics的Ebanga(ansuvimab-zykl)治疗成人和儿童(包括由扎伊尔埃博拉病毒感染RT-PCR检测阳性母亲所生的新生儿)的扎伊尔埃博拉病毒感染。这是继再生元公司的中和抗体鸡尾酒疗法Inmazeb(atoltivimab、maftivimaba and odesivimab-ebgn)在2020年10月获批上市之后,FDA批准的第二款治疗埃博拉病毒感染的药物。Ebanga也是唯一一个获得FDA批准的可冷冻干燥的埃博拉治疗单剂,并被美国FDA授予了孤儿药和突破性疗法的称号。
Ebanga是一种单克隆抗体,是从刚果民主共和国城市基威特市1995年埃博拉疫情幸存者中分离出来的。Ebanga阻断病毒与细胞受体的结合,阻止其进入细胞。2018~2019年刚果民主共和国爆发埃博拉疫情期间,Ebanga在一项临床试验(PALM试验)中,其安全性和有效性在多中心,开放标签,随机对照试验中进行了评估。174名确诊埃博拉病毒感染的参与者(120名成人和54名儿科患者)接受Ebanga单次50毫克/千克静脉输注,168名参与者(135名成人和33名儿科患者)接受调查对照。主要疗效终点为28天死亡率。主要分析人群是所有在试验的同一时间段内同时接受Ebanga或研究对照的随机患者。在接受Ebanga治疗的174例患者中,35.1%在28天后死亡,而在接受对照治疗的168例患者中,49.4%在28天后死亡。
Blueprint的Ayvakit成为美国首个获批治疗罕见胃肠道间质瘤突变的靶向疗法
2020年1月9日,美国FDA宣布批准Blueprint Medicines公司开发的Ayvakit(avapritinib)上市,用于治疗血小板衍生生长因子受体α(PDGFRA) 外显子18突变(包括D842V 突变)的不可切除性或转移性的成人胃肠道间质肿瘤(GIST)。
GIST是一种罕见的由基因组驱动的胃肠道肉瘤,其起源于胃肠道间叶组织的肿瘤,占消化道间叶肿瘤的绝大部分。数据显示,高达85%的GIST肿瘤存在PDGFRA和/或KIT基因突变,现有药物(如伊马替尼、舒尼替尼)能够结合其异常蛋白抑制相关活性,改善疾病进展。然而,约6%的新诊断GIST患者具有PDGFRA外显子18突变(其中D842V突变最为常见),其对所有已批准的疗法耐药。
Ayvakit是目前FDA批准的第一个GIST精准疗法,也是唯一一个对PDGFRA基因18号外显子突变型GIST具有高活性的药物,可以选择性强效结合PDGFRA(包括外显子18突变)和KIT突变激酶。其作为一种I型抑制剂,Ayvakit可以靶向活性激酶构象,抑制致癌激酶的信号转导,进而抑制癌症的发生发展。临床结果显示,PDGFRA外显子18突变的受试者中,总体响应率(ORR)达到84%,其中完全缓解(CR)为7%,部分缓解(PR)为77%;在PDGFR-αD842V突变的患者亚组中,ORR为89%,CR和PR分别为8%和82%;61%的外显子18突变应答患者应答时间持续了六个月或更长时间,展现了长效的治疗作用。
Epizyme 公司Tazverik美国获批治疗上皮样肉瘤
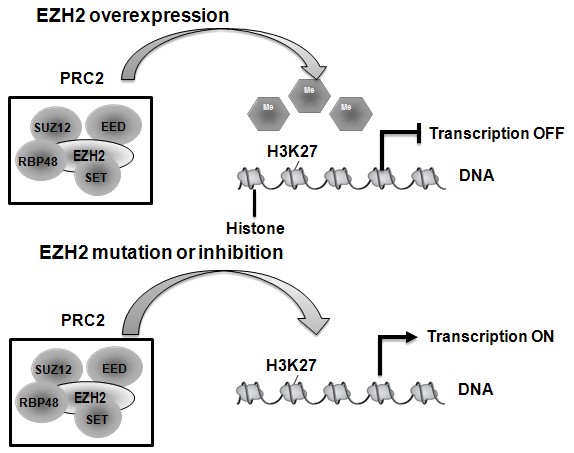
2020年1月23日,美国FDA宣布批准Epizyme 公司开发的EZH2抑制剂Tazverik(tazemetostat)上市,用于治疗不适合手术的、转移性或局部晚期成人或儿童上皮样肉瘤(ES)患者。
ES是临床上较为见的上皮样软组织肿瘤,其组织来源不明,可能是起源于具有多向分化潜能的原始间叶细胞的肿瘤。上皮样肉瘤好发于20-40岁青壮年,男性多见,根据发病部位分为远端型和近端型,以前者多见。远端型好发于四肢末端,表现为生长缓慢的结节或斑块,近端型发生于头颈部及躯干,表现为深部多发软组织肿块。研究表明,90%以上的ES患者伴有INI1蛋白缺失,从而导致EZH2酶活度活跃,促进癌细胞的恶性增生。
|
|
|
|
|
数据来源:Nacture,兴业证券经济与金融研究院整理
|
Tazverik是肉瘤首款获批的表观遗传药物,其作为一种EZH2抑制剂,能够重组异常细胞的生长通路,促进癌细胞死亡、分化,进而缩小肿瘤。临床结果显示,Tazverik的ORR为15%,其中CR为1.5%,且有67%的患者持续反应时间(DOR)达到6个月及以上。
治疗慢性便秘,美国FDA批准Braintree实验室口服乳糖醇疗法Pizensy
2020年2月12日,美国FDA宣布批准
Braintree
Laboratories开发的口服乳糖醇疗法Pizensy(lactitol)上市,用于治疗慢性特发性便秘(CIC)成人患者。
CIC是一种非常常见的疾病,主要症状是长期排便困难和不频繁,其它症状包括腹痛和腹胀。据统计,美国有3500万CIC患者,接近八分之一的美国人受到这一症状的困扰。很多患者使用包括泻药在内的不同处方或非处方药进行治疗,尽管有些患者的症状得到缓解,但药物的依赖性也给患者带来了很大的困扰。
Pizensy是一种口服的乳果糖类似物。它是一种渗透性泻药(osmotic laxitaive),可促进水分流入肠道,进而在结肠内达到通便作用。临床结果显示,与安慰剂组相比,治疗组中 25%的患者在给定的一周内至少达到了3次完全自发排便(CSBMs),或较基线时至少增加1次CSBMs,而安慰剂组中达到这一标准的患者比例为 13%。
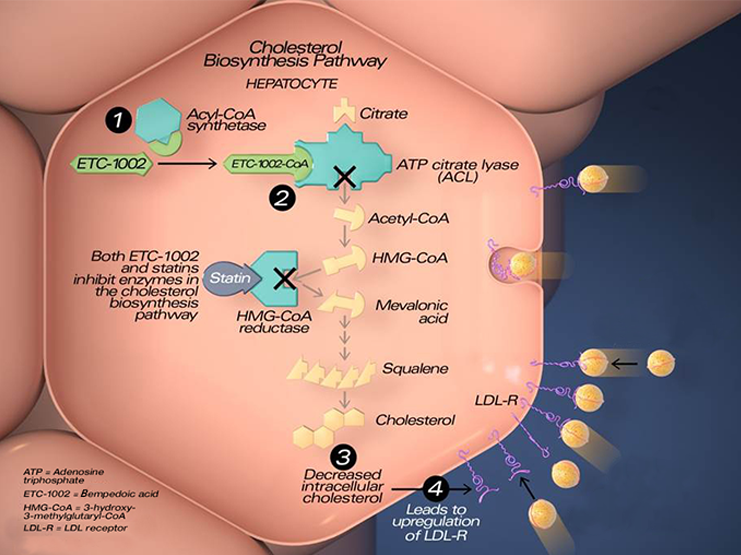
Esperion Therapeutics口服非他汀类降胆固醇新药Nexletol获FDA批准
2020年2月21日,美国FDA宣布批准
Esperion Therapeutics
公司开发的降胆固醇新药Nexletol(bempedoic acid)上市,作为饮食和最大耐受剂量他汀类药物的辅助疗法,用于治疗杂合子家族性高胆固醇血症(HeFH)成人患者,以及需要进一步降低低密度脂蛋白胆固醇(LDL-C)水平的动脉粥样硬化性心血管疾病(ASCVD)成人患者。
LDL-C是一种存在于人体内的蜡状脂肪样物质。升高的LDL-C会促进动脉中LDL-C的堆积,并可能导致心血管事件,包括心脏病发作和中风。尽管接受标准的护理治疗,包括他汀类药物治疗,但据估计,美国有近1500万患者(约四分之一的患者)不能达到指南推荐的LDL-C水平。
Nexletol是近20年来在美国批准的第一种口服、每日一次、非他汀类降胆固醇药物,在降低LDL-C方面具有一种全新的作用机制。其活性药物成分为bempedoic acid,这是一种首创(first-in-class)ATP柠檬酸裂解酶(ACL)抑制剂,通过降低胆固醇生物合成和上调LDL受体来降低LDL-C。bempedoic acid作为一种人工合成的二羧酸衍生物,是一种前体药物,需要极长链乙酰辅酶A合成酶1(ACSVL1)的激活,无法在缺乏相关酶的骨骼肌激活,可避免他汀类药物相关的肌肉毒性。临床结果显示,当与最大耐受剂量他汀类药物联合用药时,与安慰剂相比,Nexletol将LDL-C水平降低18%。在他汀类药物不耐受的患者中,与安慰剂相比,Nexletol将LDL-C水平降低28%。此外,在治疗第12周,Nexletol还将高敏C反应蛋白(Hs-CRP)水平相对基线降低19%-22%,这是心血管疾病相关炎症的关键标志物。在伴有糖尿病的患者中,与安慰剂相比,Nexletol将糖化血红蛋白(HbA1c)降低了0.2%。
术后恶心呕吐新药:美国FDA批准Acacia Pharma的Barhemsys上市
2020年2月26日,美国FDA宣布批准Acacia Pharma公司开发的Barhemsys(amisulpride)上市,用于预防和治疗术后恶心呕吐(PONV)的成人患者。该批准涵盖了已使用一种不同类别的止吐药预防性治疗PONV的手术患者或没有接受止吐药预防性治疗PONV的手术患者,以及单独使用或与不同类别的止吐药联合使用预防PONV。
PONV在手术患者中仍是一个重大问题,过去20年里相关治疗的进展甚微。PONV常被认为是手术中最不受欢迎的并发症,甚至比疼痛还要严重。目前,PONV治疗主要为预防性,而治疗失败的患者,尚无批准的有效抢救性治疗方案。在美国,据估计每年有1600万手术患者尽管接受了预防性治疗仍会发生PONV。
在接受当前标准护理预防性治疗PONV失败的患者中,Barhemsys是第一个也是唯一一个被批准用于抢救性治疗PONV的止吐药。Barhemsys是氨磺必利(amisulpride)的静脉制剂(2.5mg/mL)。氨磺必利是一种选择性多巴胺D2和D3受体拮抗剂,其片剂用于治疗精神分裂症。D2受体位于化学感受器触发区(CTZ),对从神经末梢释放的多巴胺有反应。CTZ的激活将刺激传递给呕吐所涉及的呕吐中心。对多种物种的研究表明,在视网膜后区域中的D3受体在呕吐中也起作用。临床结果显示,常用止吐药预防性治疗失败的PONV患者中,经10 mg组Barhemsys抢救的患者,呕吐缓解比例为42%,安慰剂组仅为29%。此外,Barhemsys联用其他止吐药能够显著提高PONV的保护作用,呕吐缓解比例为58%,安慰剂组仅为47%。
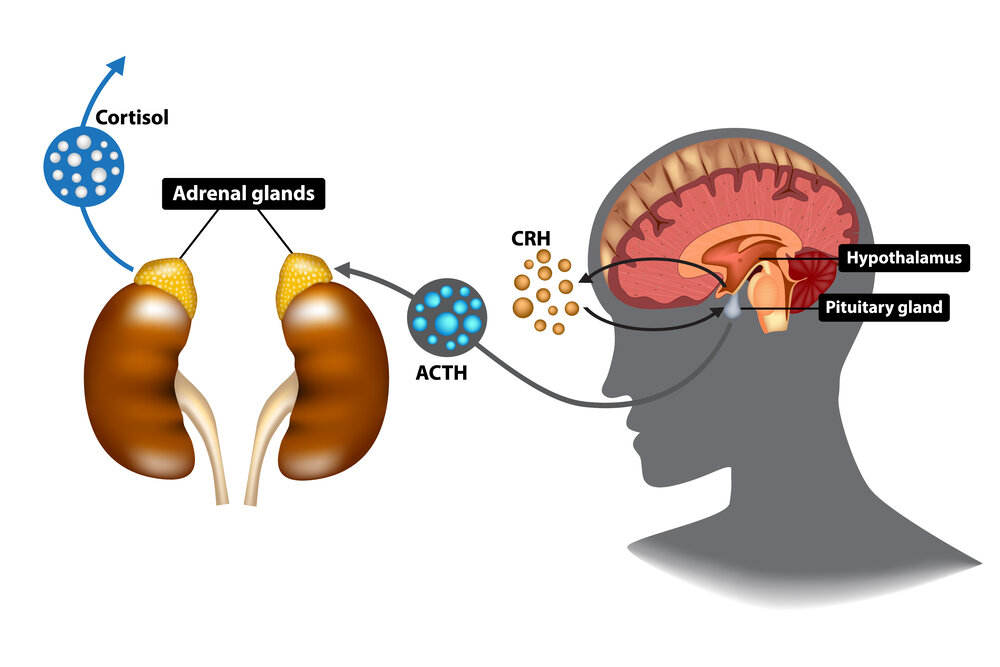
Recordati公司皮质醇合成抑制剂Isturisa获FDA批准治疗库欣综合征
2020年3月6日,美国FDA宣布批准Recordati公司开发的Isturisa(osilodrostat)上市,用于治疗成人内源性库欣综合征(Cushing’ s syndrome,CS)。
CS又称皮质醇增多症(hypercortisolism),是由于机体多种病因引起的,肾上腺皮质长期分泌过多糖皮质激素(主要为皮质醇)所产生的一系列的临床表现,也称为内源性库欣综合征。其按病因可分为促肾上腺皮质激素(ACTH)依赖型和非依赖型两种,主要表现为满月脸、多血质外貌、向心性肥胖、痤疮、紫纹、高血压、继发性糖尿病和骨质疏松等,高发年龄在20~40岁,男女发病率之比约为1:3。
Isturisa的活性药物成分为osilodrostat,是一种皮质醇合成抑制剂。Isturisa是第一种通过FDA批准的药物,通过抑制11-β羟化酶发挥作用,这种酶在肾上腺皮质醇生物合成的最后一步发挥作用。临床结果显示,Isturisa维持治疗的患者中,86%的患者皮质醇水平在正常范围内,而服用安慰剂的患者组的数值为30%。在接受治疗24周之后,接近一半患者的皮质醇水平降低到正常范围内。
百时美施贵宝多发性硬化症新药Zeposia获美国FDA批准
2020年3月25日,美国FDA宣布批准百时美施贵宝(BMS)开发的口服新药S1P受体调节剂Zeposia(ozanimod)上市,用于治疗成人复发型多发性硬化症(RMS),包括临床孤立综合征、复发缓解性疾病、活动性继发进展性疾病。
多发性硬化(MS)是一种免疫系统攻击覆盖神经的保护性髓鞘的疾病,能够造成损害性损伤,使信号更难在每个神经细胞之间传播。这种“信号崩溃”可导致疾病症状和复发。MS目前影响全球230万人,受影响的女性多于男性。该病特征是脱髓鞘和轴突丢失,导致神经功能受损和严重致残。MS的主要亚型是复发型多发性硬化症(RMS),占MS患者的85%,包括临床孤立综合征(CIS)、复发-缓解型多发性硬化症(RRMS)和活动性继发性进行性多发性硬化症(SPMS)。
Zeposia是一种口服药物,是唯一被批准具有以下特征的鞘氨醇-1-磷酸(S1P)受体调节剂:在RMS患者启动治疗时,无需进行基因测试、无需基于标签的首次剂量观察。Zeposia可以与淋巴细胞表面的S1PR1和S1PR5具有较高的亲和力,当其与受体结合后就可以阻止淋巴细胞离开淋巴结进入中枢神经系统,发挥抗炎作用。此外,Zeposia也能进入中枢神经系统,直接与少突胶质细胞和星形胶质细胞上的S1PR结合,促进髓鞘再生和防止炎症。Zeposia与Avonex(干扰素β-1a)的头对头临床结果显示,治疗一年后,Zeposia组相比Avonex组ARR降低48%,治疗两年后ARR降低38%。此外,治疗一年后,Zeposia相比Avonex将T1加权钆增强脑损伤相对减少68%,T2损伤的数量相对减少了48%;治疗两年后,Zeposia相比Avonex将T1加权钆增强脑损伤相对减少53%,T2损伤的数量相对减少了42%。
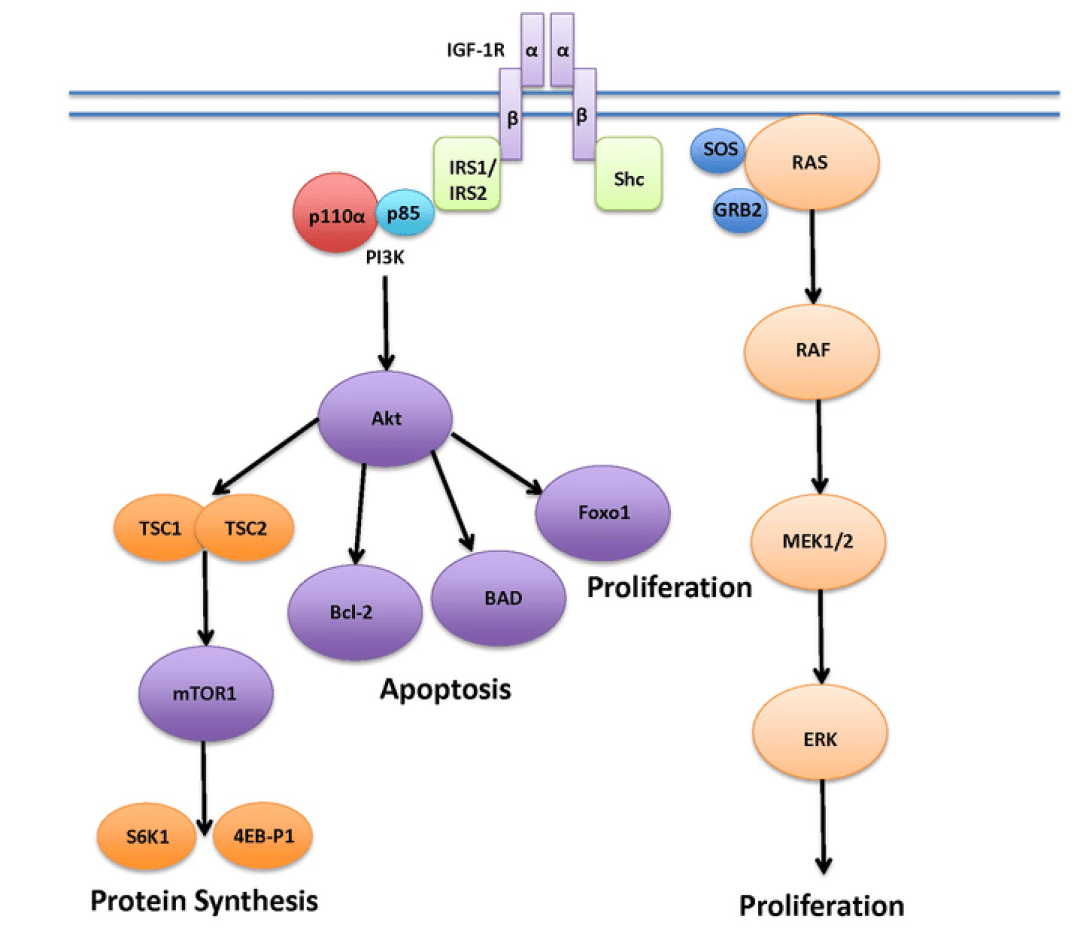
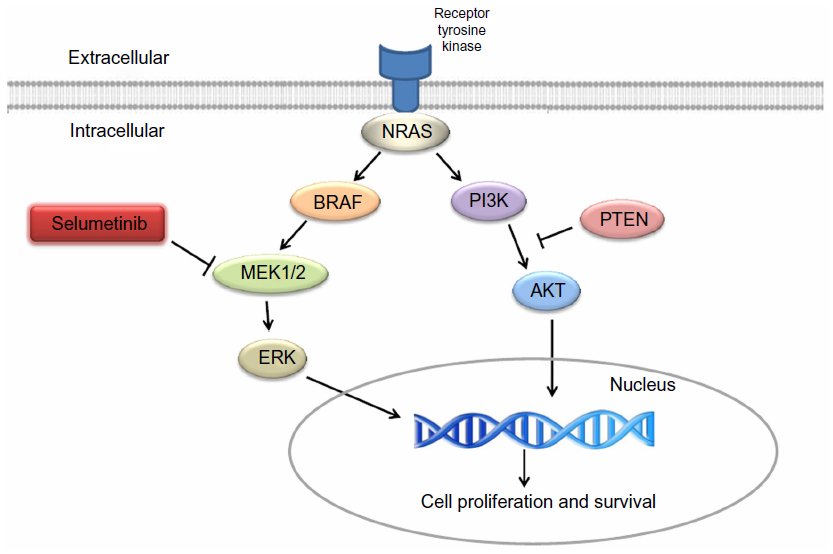
2020年4月10日,美国FDA宣布批准阿斯利康和默沙东共同开发的Koselugo(selumetinib)司美替尼上市,用于治疗2岁及2岁以上的1型神经纤维瘤病(NF1)儿童患者,这些患者携带有表现出症状和/或进行性,不能通过手术治疗的丛状神经纤维瘤(PN)。
NF1是一种罕见、不可治愈的遗传性疾病,其是因为合成神经纤维瘤蛋白(neurofibromin)的NF1基因发生突变引起的。该基因突变可扰乱RAS/MAPK信号通路(RAS-RAF-MEK-ERK),进而导致肿瘤的生长。NF1的症状包括皮肤神经纤维瘤、皮肤色素沉积。在20-50%的NF1患者中,肿瘤在神经鞘上发展,导致PN。这些PNs可导致疼痛,运动功能障碍,气道功能障碍,肠/膀胱功能障碍和毁容,并有可能转变为恶性外周神经鞘瘤(MPNST)。
Koselugo是首款获得FDA批准治疗这种在生命早期就使人衰弱的罕见疾病药物,是一种能够靶向MEK的激酶抑制剂。MEK是RAS/MAPK信号通路中的关键蛋白激酶,Koselugo能够选择性抑制MEK1和MEK2,从而使失调的信号通路恢复正常,进而缓解儿童NF1患者的病情。实验结果显示,Koselugo的治疗使患者的总缓解率(ORR)达到66%,所有患者均为部分缓解。在这些患者中,82%的患者缓解持续时间达到了12个月或更长。
FDA批准Seattle Genetics的口服酪氨酸激酶抑制剂Tukysa治疗HER2阳性转移性乳腺癌
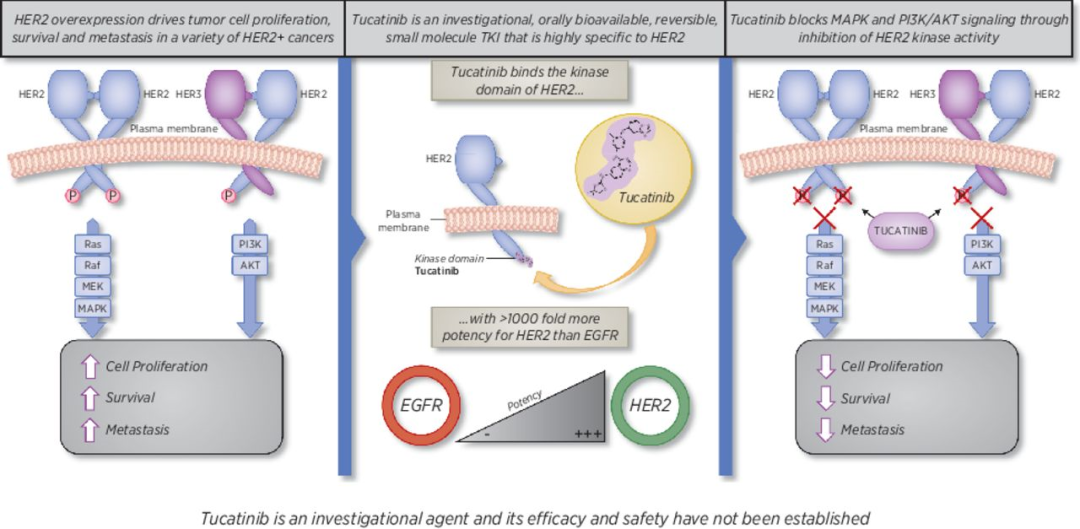
2020年4月17日,美国FDA宣布批准Seattle Genetics公司开发的口服酪氨酸激酶抑制剂Tukysa(tucatinib)上市,与曲妥珠单抗和卡培他滨联合,用于治疗不能手术切除或转移性HER2阳性乳腺癌成年患者。
HER2阳性乳腺癌约占全部乳腺癌的20-25%,这类患者体内表达过多的人表皮生长因子受体2(HER2)蛋白质,其会促进癌细胞的生长。因此,这种类型的乳腺癌侵袭程度较高、预后差,并且超过25%的患者会发生脑转移。目前,乳腺癌处于单靶点靶向治疗时代,曲妥珠单抗已显著提高HER2阳性早期乳腺癌患者的生存率,但仍有部分高危患者对药物没有响应或者用药后产生了耐药性,临床治愈的需求尚无法满足。
Tukysa是首个基于国际合作试点项目Orbis而批准的新分子实体(NME)药物,能够特异性抑制HER2蛋白。临床结果显示,与安慰剂+曲妥珠单抗+卡培他滨方案相比,Tukysa+曲妥珠单抗+卡培他滨方案显著延长了无进展生存期(7.8个月vs 5.6个月)、总生存期(21.9个月vs 17.4个月),试验组脑转移患者的平均中位无进展生存期为7.6个月,而对照组患者则为5.4个月。
Incyte公司Pemazyre美国获批二线治疗晚期胆管癌
2020年4月17日,美国FDA宣布批准Incyte公司开发的Pemazyre(pmigatinibe)上市,用于治疗接受过治疗的、无法切除的局部晚期或转移性胆管癌的成年患者。
胆管癌是在胆管中形成的罕见癌症,其根据解剖起源可分为肝内胆管癌(iCCA)和肝外胆管癌,通常在预后较差时被诊断为晚期。 胆管癌的发病率因地区而异,在北美和欧洲,每10万人的发生率在0.3-3.4之间。成纤维细胞生长因子受体(FGFR2)的融合或重排几乎仅发生在iCCA中,能够在10-16%的患者中观察到。FGFR在肿瘤细胞的增殖,存活,迁移和血管生成中起重要作用。FGFRs中的激活融合,重排,易位和基因扩增与各种癌症的发生密切相关。
Pemazyre是FGFR1/2/3的激酶抑制剂,是该适应症的首个也是唯一的FDA批准的治疗方法。临床结果显示,在携带FGFR2融合或重排的患者中,Pemazyre单药治疗的总缓解率为36%,中位DOR为9.1个月。Pemazyre获得了FDA授予的用于胆管癌的治疗孤儿药称号。
帕金森病新药:Neurocrine Biosciences第三代强效COMT抑制剂Ongentys获美国FDA批准
2020年4月24日,美国FDA宣布批准
Neurocrine Biosciences
公司开发的Ongentys(opicapone)上市,作为左旋多巴/卡比多巴的一种辅助疗法,用于治疗正在经历运动波动(OFF)期的帕金森病(PD)患者。
帕金森病是一种慢性神经退行性疾病,在全球约有600万名患者。帕金森病目前还没有治愈的方法,患者通常需要使用左旋多巴治疗,替代缺失的多巴胺。长期使用这种药物会导致严重的运动能力波动。这种波动分为两个阶段——在“开启”期,患者的运动能力正常;而在“关闭”期,患者的运动能力明显下降,通常表现为运动缓慢、僵硬、行走障碍、震颤和姿势不稳定,极大地影响了患者的日常生活。
Ongentys是一种每日一次的口服选择性儿茶酚-氧位-甲基转移酶(COMT)抑制剂,是第一个也是唯一一个被批准的每日一次COMT抑制剂。Ongentys通过阻断分解左旋多巴的COMT酶,减少左旋多巴在血液中的分解从而保护左旋多巴,使更多的左旋多巴可以到达大脑,延长其临床效果,帮助患者实现运动症状控制。临床结果显示,在正经历“OFF”期的帕金森病患者中,与安慰剂组相比,Ongentys加用左旋多巴/卡比多巴,可减少“OFF”期,同时增加“ON”期,并且不会出现令人烦恼的运动障碍。同时,在“ON”期,帕金森病患者的运动症状能得到更好的控制。
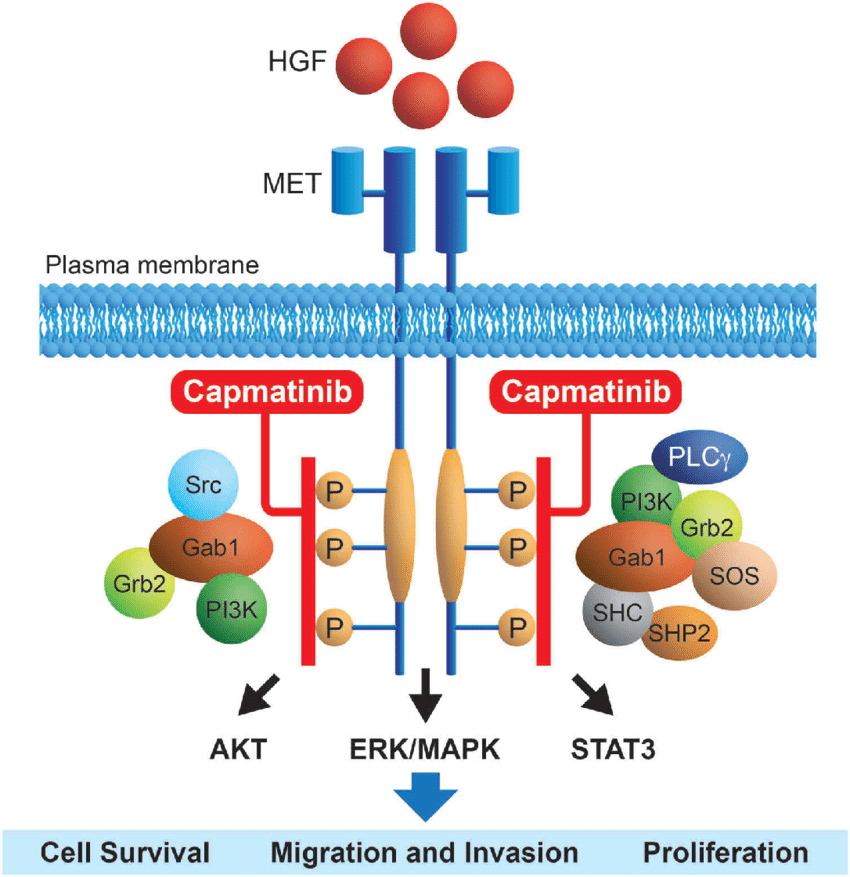
美国首个METex14突变肺癌靶向抗癌药:诺华强效MET抑制剂Tabrecta获FDA批准
2020年5月6日,美国FDA宣布批准诺华(Novartis)公司开发的Tabrecta(capmatinib)上市,用于治疗携带MET外显子14跳跃(MET exon14 skipping,METex14)突变的转移性非小细胞肺癌(NSCLC)成人患者,包括一线治疗(初治)患者和先前接受过治疗(经治)的患者。
肺癌是最常见的癌症,全球每年确诊约200万新病例,美国约22.8万例。非小细胞肺癌(NSCLC)是最常见的肺癌类型,约占所有肺癌病例的85%。约70%的NSCLC患者携带基因组突变。METex14突变是一种公认的致癌驱动因素,发生在3-4%的新诊转移性NSCLC病例中。虽然罕见,但这种突变是预后不良的指标,且在Tabrecta获批之前没有专门针对METex14突变转移性NSCLC的治疗方法。
Tabrecta是第一个也是唯一一个被FDA批准专门针对METex14突变转移性NSCLC的疗法,包括用于初治患者以及经治患者,无论先前治疗药物类型如何。Tabrecta作为一种口服MET抑制剂,能够阻断癌症发生发展中的关键激酶,控制癌症进展。临床结果显示,在初治患者中,Tabrecta的ORR为68%,中位缓解时间DoR为12.6个月;在经治患者中,ORR为41%,DoR为9.7个月。
|
|
|
|
|
数据来源:PubMed,兴业证券经济与金融研究院整理
|
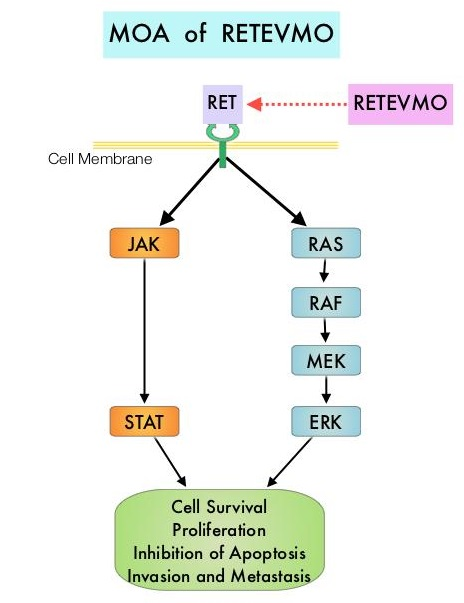
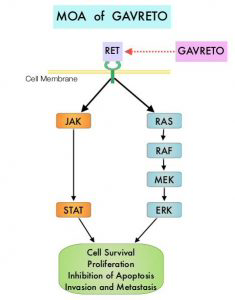
全球首个RET激酶抑制剂:礼来“不限癌种”靶向抗癌药Retevmo获美国FDA批准
2020年5月8日,美国FDA宣布批准礼来子公司Loxo Oncology开发的靶向抗癌药Retevmo(selpercatinib)上市,用于治疗肿瘤在特定基因(转染期间重排基因,RET基因)存在基因改变(突变或融合)的3种类型肿瘤患者:非小细胞肺癌(NSCLC)、甲状腺髓样癌(MTC)、其他类型的甲状腺癌。
RET基因是一个在转染过程中发生重排的原癌基因,并因此而得名,该基因编码一种细胞膜受体酪氨酸激酶,其异常是多种类型肿瘤的罕见驱动因素。据估计,RET融合存在于大约2%的非小细胞肺癌(NSCLC)、10-20%的乳头状甲状腺癌(PTC)和其他类型甲状腺癌;RET点突变存在于大约60%的甲状腺髓样癌(MTC)中。RET融合及RET点突变癌症主要依赖于RET激酶的激活来维持其增殖和存活,这种依赖性通常被称为“致癌基因成瘾”,使得这类肿瘤对靶向RET的小分子抑制剂高度敏感。
Retevmo是第一个被批准专门用于携带RET基因改变的癌症患者的治疗药物,是一种强效、口服、高度选择性、转染期间重排(RET)激酶抑制剂,旨在抑制天然的RET信号转导以及预期的获得性耐药机制,开发用于治疗肿瘤中携带异常的RET激酶的患者。临床结果显示,对于NSCLC治疗,经治患者ORR为64%,81%的应答患者DoR达6个月及以上;初治患者ORR为84%,58%的应答患者DoR达6个月以上;此外,高达50%的RET融合阳性NSCLC患者可能存在肿瘤脑转移,在存在基线脑转移的患者中,Retevmo显示出强劲疗效,颅内缓解(CNS-ORR)高达91%(n=10/11)。对于MTC治疗,经治患者ORR为69%,76%的应答患者DoR达6个月及以上;初治患者ORR为73%,61%的应答患者DoR达6个月以上。对于RET融合阳性甲状腺癌的治疗,放射性碘难治且接受过另一系统疗法的患者ORR为79%,87%的应答患者DoR达6个月及以上;仅接受过放射性碘治疗的患者ORR为100%,75%的应答患者DoR达6个月以上。
Deciphera Pharmaceuticals胃肠道间质瘤靶向药Qinlock在美国批准上市
2020年5月15日,美国FDA宣布批准
Deciphera Pharmaceuticals
公司开发的蛋白激酶抑制剂Qinlock(ripretinib)片剂上市,用于治疗已经接受过3种以上蛋白激酶抑制剂疗法的晚期胃肠道间质瘤(GIST)成年患者。
GIST是一种由基因突变驱动的胃肠道肉瘤,源于胃肠道壁中的特殊神经细胞。患者中最常见的突变为KIT蛋白激酶突变,大约占80%的病例。大约6%的新确诊患者携带PDGFRα突变。
Qinlock是第一个被批准用于四线治疗GIST的药物。其活性药物成分为ripretinib,是一种KIT/PDGFRα激酶开关调控抑制剂,用于治疗KIT/PDGFRα驱动的胃肠道间质瘤(GIST)、系统性肥大细胞增多症(SM)以及其他癌症。ripretinib通过抑制KIT和PDGFRα的广谱突变来改善胃肠道间质瘤患者的治疗,可阻断胃肠道间质瘤中涉及第9、11、13、14、17、18外显子中的起始和继发KIT突变以及SM中发现的原发性KIT第17号外显子D816V突变。此外,ripretinib还能够抑制第12、14、18外显子中的原发性PDGFRα突变,包括涉及第18外显子D842V突变的胃肠道间质瘤。临床结果显示,与安慰剂组相比,Qinlock治疗组无进展生存期显著延长(中位PFS:6.3个月 vs 1.0个月)、疾病进展或死亡风险显著降低85%。总生存期(OS)方面,Qinlock治疗组与安慰剂组相比显著延长(中位OS:15.1个月 vs 6.6个月)、死亡风险降低64%,且安慰剂组的OS数据包括接受安慰剂治疗病情进展后转向Qinlock治疗的患者数据。总缓解率(ORR)方面,Qinlock治疗组与安慰剂组相比大幅提高(ORR:9.4% vs 0%)。
FDA批准Zionexa公司的放射性诊断剂Cerianna上市
2020年5月20日,美国FDA宣布批准Zionexa公司开发的放射性诊断剂市Cerianna(fluoroestrdiol F18)上市,作为活检的辅助手段与正电子发射断层扫描(PET)成像共用,检测雌激素受体(ER)阳性的复发或转移性乳腺癌患者。
CERIANNA是一种无菌、澄清、无色的静脉注射溶液,其活性成分氟雌二醇F18(fluoroestrdiol F18)是一种合成的雌激素类似物。一项纳入了90名经组织学证实为浸润性乳腺癌的女性的临床试验数据为此次批准提供支持。数据结果通过评分方式得出结果,在47例活检阳性患者中,有36例影像学检查阳性;在38例活检阴性患者中,所有38例影像学均为阴性;11例影像学检查假阴性的患者中,有10例活检阳性。
Amivas公司抗疟药Artesunate在美获批上市
2020年5月26日,美国FDA宣布批准Amivas公司开发的青蒿琥酯(Artesunate)静脉注射剂上市,用于治疗患有严重疟疾的成人和儿童。
疟疾是一种由蚊子叮咬传播的寄生虫病,其流行程度和危险性丝毫不亚于癌症等其他重大疾病。疟疾一旦发展严重,即会影响到中枢神经系统。据WHO估计,2018年,全球疟疾病例数超过2.2亿,约造成40.5万人死亡。
青蒿琥酯是目前美国唯一一款批准用于治疗严重疟疾的药物。Artesunate的安全性和有效性在两项临床试验中得到评估:亚洲进行的一项随机对照试验以及在非洲开展的一项支持性随机对照试验中,使用Artesunate治疗的患者在院内死亡的人数均明显低于使用奎宁治疗的对照组患者。
阿尔茨海默病诊断技术新发现:礼来Tauvid获FDA首批
2020年5月28日,美国FDA宣布批准礼来公司开发的放射性诊断剂Tauvid(flortaucipir F18)上市,用于大脑的正电子发射断层扫描(PET)成像,以评估脑中聚集的tau神经原纤维缠结(NFT)的密度和分布。
阿尔茨海默病是一种渐进性疾病,最初的症状为轻度记忆丧失。Tau神经纤维缠结和淀粉样蛋白沉积被认为是阿尔茨海默病的标志。在阿尔茨海默病患者中,大脑中的神经元内部会出现病理形态的tau蛋白,形成神经纤维缠结。
Tauvid是一种放射性诊断试剂,是第一种获得FDA批准,帮助对大脑中的tau病理进行成像的药物。Tauvid经静脉给药后,在大脑中与tau蛋白错误折叠积累的区域结合,通过使用PET扫描对大脑进行成像,可以帮助识别是否存在tau蛋白病理。临床结果显示,解读Tauvid图像的评估者有较高几率正确判断存在tau病理的患者,评估无tau病理患者的准确率为中等到高。另一项研究检测了Tauvid评估者的解读与其他评估结果之间的一致性,将解读完全吻合的一致性为1,而完全不吻合的一致性为0。在这一研究中,对所有241例患者的解读一致性为0.87。亚组分析表明,82例终末期疾病患者的一致性为0.82,159名认知功能障碍患者的一致性为0.90。
FDA批准Jazz Pharmaceuticals小细胞肺癌新药Zepzelca
2020年6月15日,美国FDA宣布批准Jazz Pharmaceuticals及其合作伙伴PharmaMar公司开发的Zepzelca(lurbinectin)上市,用于治疗铂类化疗中或化疗后疾病进展的转移性小细胞肺癌(SCLC)成年患者。
SCLC是一类神经内分泌肿瘤,占肺癌的15%。SCLC是与更为常见的NSCLC完全不同的肿瘤,其分子变异、增长速度、转移速度都有显著差异。SCLC虽然对化疗非常敏感,但其复发率相对较高,复发后成为一类难治的肿瘤。
Zepzelca是一种海鞘素衍生物,是1996年以来第一个被批准用于二线治疗SCLC的新药。其作为一种RNA聚合酶II的抑制剂,通过阻滞RNA聚合酶II与DNA的结合,并降解RNA聚合酶II的催化亚基RPB1,从转录的起始至延长阶段发挥抑制转录活性,使肿瘤细胞在有丝分裂过程中畸变、凋亡、最终减少细胞增殖。临床结果显示,Zepzelca单药治疗复发性SCLC的ORR为35%、中位DoR为5.3个月;独立审查委员会(IRC)评估的ORR为30%、中位DoR为5.1个月。
|
|
|
|
|
数据来源:PubMed,兴业证券经济与金融研究院整理
|
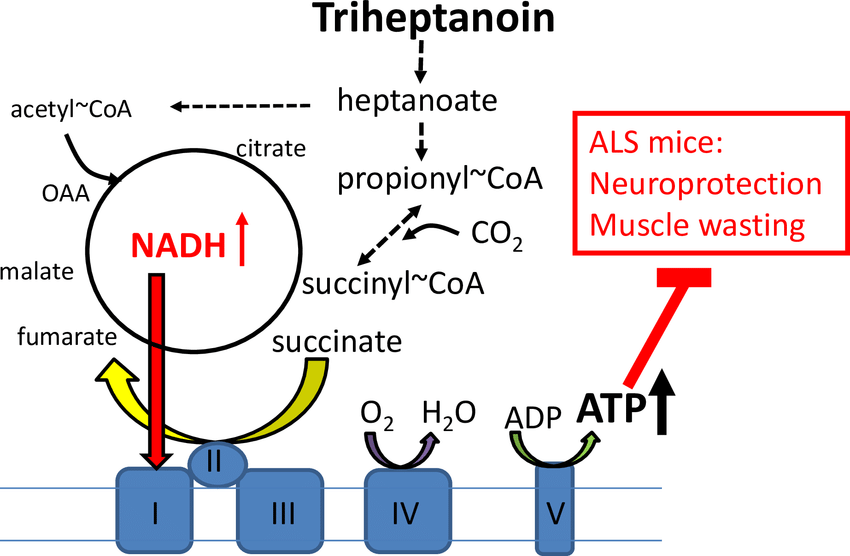
Ultragenyx Pharmaceutical公司Dojolvi美国获批治疗长链脂肪酸氧化障碍
2020年6月30日,美国FDA宣布批准Ultragenyx Pharmaceutical公司开发的Dojolvi(triheptanoin)三庚酸甘油酯上市,作为卡路里和脂肪酸的来源,用于治疗通过分子诊断确诊的长链脂肪酸氧化障碍(LC-FAOD)儿童和成人患者。
LC-FAOD是一组以机体不能将长链脂肪酸转化为能量的代谢缺陷为特征的常染色体隐性遗传病。患者由于不能将脂肪转化为能量,导致体内葡萄糖严重耗竭,出现严重并发症,从而导致住院或早夭。由于存在包括早期死亡在内的严重结局风险,LC-FAOD被纳入美国和某些欧洲国家的新生儿筛查检测。
Dojolvi(triheptanoin)三庚酸甘油酯,是一种高纯度、人工合成的7碳脂肪酸甘油三酯,在甘油骨架上通过多个化学合成步骤添加了三个7碳脂肪酸形成。专门为LC-FAOD患者提供中链、奇数碳脂肪酸作为能源和代谢产物替代品。Dojolvi的批准基于多种证据的支持,其中包括包含29名患者的2期临床试验结果,包含75名患者的长期安全性和疗效扩展研究,以及20名同情用药患者和67名通过扩展使用(expanded access)接受治疗的患者的数据。
Acacia Pharma公司Byfavo美国获批诱导和维持接受医疗程序过程中镇静
2020年7月2日,美国FDA宣布批准Acacia Pharma公司开发的Byfavo(Remimazolam)上市,用于诱导和维持持续30分钟或更短程序的成年人的程序镇静作用。
Byfavo是一种非常快速的起效/抵消性IV苯二氮卓类镇静剂,用于持续30分钟或更短的侵入性医疗程序。其作为一种GABA激动剂,可作用于GABA受体,特别是GABA-α受体。Byfavo的安全性在包含969名患者的3个关键性研究中得到证实,他们接受了结肠镜或支气管镜检查,其中630名患者使用了Byfavo。临床结果显示,最常见的不良事件为低血压和高血压。
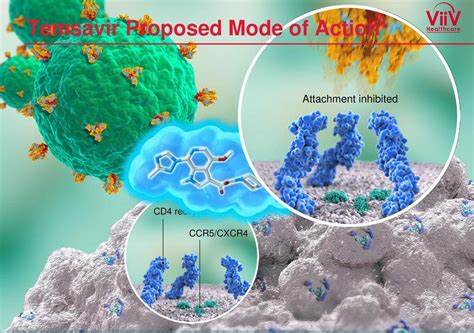
抗HIV重磅:葛兰素史克首创附着抑制剂Rukobia获美国FDA批准
2020年7月2日,美国FDA宣布批准ViiV Healthcare公司开发的Rukobia(fostemsavir)上市,用于治疗曾尝试过多种HIV疗法(heavily treatment-experienced,HTE)、并且由于耐药/不耐受或安全性的考虑而对其当前ARV方案治疗失败的多重耐药HIV-1成人感染者。
在过去30年里,HIV治疗方面取得了令人难以置信的进步。抗逆转录病毒药物能够有效抑制HIV,这有助于减少疾病进展、HIV传播和与艾滋病有关的死亡。由于HIV具有不断变化的能力,一些患者可能对抗逆转录病毒药物产生耐药性,导致其治疗方案失败。在耐受性、安全性和药物相互作用方面受到的挑战,可能会进一步减少在设计有效治疗方案时可接受的抗逆转录病毒疗法的数量。对于既往接受过多种方案并且无法成功抑制HIV的多重耐药患者群体而言,仍然存在着显著未得到满足的医疗需求。
Rukobia的活性药物成分为fostemsavir,这是一种首创的(first-in-class)HIV-1附着抑制剂。fostemsavir是temsavir的一种前药,通过口服后,fostemsavir可转变为temsavir,然后被吸收并通过直接附着在病毒表面糖蛋白120(gp120)亚基发挥抗病毒作用。通过与病毒上的这个位置结合,temsavir可阻止HIV病毒附着到宿主免疫系统CD4+T细胞和其他免疫细胞上,并防止HIV病毒感染这些细胞并增殖。由于Rukobia是第一个针对病毒周期中第一步(附着)的抗逆转录病毒疗法,因此对其他种类的抗逆转录病毒药物没有显示出耐药性,这可能帮助对大多数其他药物产生耐药性的HIV感染者。临床结果显示,大多数(60%)HTE多重耐药HIV-1成人感染者接受Rukobia和优化的背景治疗后,实现并维持病毒抑制直至96周。
首个口服低甲基化制剂:大冢抗癌药Inqovi获美国FDA批准
2020年7月7日,美国FDA宣布批准大冢制药(Otsuka Phamra)全资子公司Astex公司开发的口服固定剂量组合抗癌药Inqovi(decitabine and cedazuridine)上市,用于治疗骨髓增生异常综合症(MDS)和慢性骨髓单核细胞白血病(CMML)成人患者。
骨髓增生异常综合征(MDS)是一组一种造血干细胞疾病,其特征是髓样、红系和巨核细胞发育异常改变。据统计,美国每年MDS发病约为10,000,其中1/3的患者可以发展成急性骨髓性白血病(AML)。一直起来,静脉注射和皮下注射低甲基化剂一直是MDS和慢性粒细胞单核细胞白血病(CMML)治疗的基石药物,比如注射用地西他滨,但是每天注射给患者带来了麻烦和痛苦。
Inqovi片剂是美国首个被批准治疗MDS和CMML的口服去甲基化制剂,其中的cedazuridine组分能够抑制肠道和肝脏中的胞苷脱氨酶,避免降解地西他滨,从而能够实现口服给药地西他滨,达到与静脉输注地西他滨同等的暴露当量。临床结果显示,静脉注射地西他滨和口服Inqovi相比,在患者体内达到的药物浓度相似。此外,大约一半以前依赖输血的患者在接受治疗8周内不再需要输血。Inqovi的安全性特征也与静脉注射地西他滨相似。
2020年7月24日,美国FDA宣布批准印度
Dr. Reddy’ s
Laboratories Ltd.公司开发的Xeglyze(Abametapir)上市,用于6个月及以上患者的头虱感染的局部治疗。
Xeglyze(Abametapir)0.74%洗护剂是一种金属蛋白酶抑制剂,该酶对于虫卵发育和若虫和成虫虱子的存活至关重要,Xeglyze可有效杀灭患者头部的虱子。需要注意的是,Xeglyze包含苯甲醇。某些患者全身性暴露于苯甲醇容易引起严重和致命的不良反应,包括新生儿和低出生体重婴儿的“喘气综合征”。“喘气综合征”的特征在于中枢神经系统抑制,代谢性酸中毒和喘气。目前并不清楚可能会发生毒性的最小剂量的苯甲醇是多少。早产和低出生体重的婴儿可能更容易发生不良反应。Xeglyze在6个月以下的小儿患者中尚未确定安全性和有效性。
拜耳Lampit获美国FDA批准上市,用于儿科恰加斯病患者
2020年8月6日,美国FDA宣布批准拜耳(Bayer)公司开发的Lampit(nifurtimox)上市,用于儿科患者(0岁至18岁以下,体重≥2.5公斤),治疗由克氏锥虫(T. cruzi)引起的恰加斯病(Chagas disease)
恰加斯病是一种传染性热带疾病,在美国影响了大约30万人。这种疾病在拉丁美洲大部分地区都是地方病,但在美国,这种病越来越引起人们的健康关注。克氏锥虫寄生于人和哺乳动物的血液和多种组织细胞内。其生活史中有锥鞭毛体与无鞭毛体两种形态。锥鞭毛体经皮肤创口感染侵入人体血液。该病也可通过母乳、胎盘、输血或食入传染性锥蝽粪便污染的食物而感染。估计在中、南美洲有2000万受染者。其中,大多数感染是无症状的。症状性病例仅显示轻微的流感样症状,这些症状在感染后直接出现。大约2个月后,恰加斯病进入了一个持续多年的慢性阶段,导致严重的器官损伤、最终导致心脏猝死。教育、早期诊断和治疗是预防恰加斯病的关键。
Lampit是一种新的、可分割、易分散的片剂,可以用手在刻痕线处分开。该片剂采用了一种新的配方,可以很容易地分散在水中,有助于可能有吞咽困难或需要半片片剂的儿科患者的给药治疗。恰加斯病的治疗只基于2种硝基杂环化合物,nifurtimox和benznidazole(苯硝唑)。迄今为止,nifurtimox仅作为120毫克的片剂提供,这很难给药,尤其是对年幼的儿童。Lampit的开发为儿童给药提供了新的方案。临床结果显示,在治疗结束后随访一年的血清学反应方面,60天Lampit方案优于历史安慰剂对照,达到了研究的主要终点。在整个研究群体中,60天方案与30天方案(未经批准的给药方案)相比在血清学反应方面具有优越性。研究中,Lampit基于体重调整的剂量治疗显示出良好的安全性。
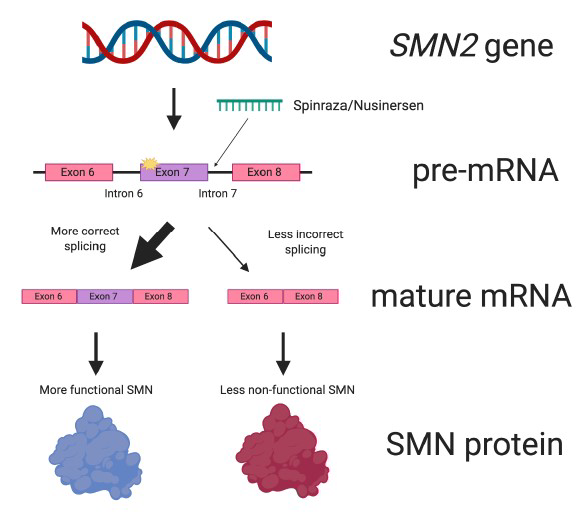
首个脊髓性肌萎缩症口服疗法罗氏Evrysdi获FDA批准
2020年8月7日,美国FDA宣布批准罗氏(Roche)旗下基因泰克公司开发的Evrysdi(risdiplam)上市,用于治疗2个月及以上儿童和成人脊髓性肌萎缩症(SMA)患者。
脊髓性肌萎缩症(SMA)是一种罕见的、进行性的遗传性神经肌肉疾病,患者由于运动神经元1(SMN1)基因突变导致SMN蛋白缺陷,患儿多表现为四肢肌肉无力、肌肉萎缩、呼吸困难等,是婴儿死亡的主要遗传原因。SMA属于罕见病,影响了万分之一的婴儿。其中,中国的SMA患者约3万人。按照发病年龄,SMA可以分为SMA-I型(6月以内发病)、II型(出生后6~18个月起病)、III型(出生18个月后发病)、IV型(成年后发病)。
Evrysdi是第一个治疗SMA的口服疗法,也是第一个可在家给药的SMA疗法。Evrysdi是一种运动神经元生存基因2(SMN2)mRNA剪接修饰剂,通过提高SMN的产生来治疗SMA。SMN蛋白遍布全身,对维持健康的运动神经元和运动至关重要。临床结果显示,接受Evrysdi治疗的婴儿,能够在没有支撑的情况下坐立至少5秒钟,这是SMA疾病自然病程中常见的关键运动里程碑。此外,与自然病史相比,Evrysdi还提高了12个月和23个月时无永久通气的生存率。
Trevena止痛创新药Olinvyk获FDA批准
2020年8月7日,美国FDA宣布批准Trevena公司开发的阿片激动剂Olinvyk(oliceridine)上市,用于治疗成人中度至重度急性疼痛,具体为:疼痛严重到需要静脉注射阿片类药物、替代疗法又不能充分控制疼痛的患者。
Olinvyk的活性药物成分为oliceridine,是一种首创的(first-in-class)静脉镇痛药。Olinvyk是首个G蛋白选择性激动剂,与静脉注射吗啡相比,镇痛效果更好。Olinvyk以μ阿片受体为靶点,通过一种优化作用机制(MOA),优先参与负责疗效的信号通路,减少引起不良反应的信号通路的激活。Olinvyk具有独特的、差异化的药代动力学(PK)特征:(1)起效迅速且疗效持久,大多数患者在给药后2-5分钟就感到疼痛缓解、疗效持续约3小时;(2)没有已知的活性代谢;(3)在老年患者或肾功能受损的患者中,无需剂量调整,将为有风险的患者提供一个新的选择。
临床结果显示,与安慰剂组相比,口服Olinvyk的患者在批准的剂量下疼痛有效减轻。需要注意的是,Olinvyk成瘾、滥用和误用会引发危及生命的呼吸抑制、新生儿阿片类戒断综合征,以及与苯二氮卓类药物或其他中枢神经系统抑制剂联合使用的风险。
NS Pharma杜氏肌营养不良新药Viltepso获FDA批准
2020年8月12日,美国FDA宣布批准日本新药株式会社(Nippon Shinyaku)子公司NS Pharma开发的Viltepso(
viltolarsen
)上市,用于治疗53号外显子跳跃(exon 53 skipping)杜氏肌营养不良(DMD)患者。
杜氏肌营养不良(DMD)是一种罕见的致命性神经肌肉遗传病,其特点是进行性肌肉退化和无力,该病是最常见的一种肌营养不良症。DMD是由DMD基因中的突变引起的,导致肌营养不良蛋白的缺失。该病最初的症状通常在3-5岁出现,并随着时间的推移而恶化。
DMD患者在2岁时就会出现进行性和不可逆的肌肉丢失症状。心脏和呼吸肌问题始于青少年时期,会导致严重的危及生命的并发症。该病是普遍致命的,患者通常在20多岁时死亡。在全世界,大约每3600名男婴中就有一名患DMD;在极少数情况下,它也会影响女性。
Viltepso是美国FDA批准的第二款治疗DMD的第53号外显子跳过疗法,是第一个也是唯一一个被证实在年龄低至4岁的儿童中可增加抗肌萎缩蛋白水平的第53号外显子跳过疗法。Viltepso是一种反义寡核苷酸药物,通过屏蔽(跳跃)抗肌萎缩蛋白(Dys)基因中的外显子53来促进功能性抗肌萎缩蛋白的产生。基于Dys水平的增加,Viltepso获得了FDA的加速批准。临床结果显示,在接受推荐剂量80mg/kg/wk(N=8)的患者中,100%的患者(8/8)在使用Viltepso治疗后显示抗肌萎缩蛋白水平增加,88%的患者(7/8)抗肌萎缩蛋白水平达到正常值的3%或更高。总体而言,在基线检查时,患者抗肌萎缩蛋白水平为正常值的0.6%,而在接受Viltepso(80 mg/kg/wk)治疗20-24周后,观察到患者抗肌萎缩蛋白水平增加到正常值的6%。
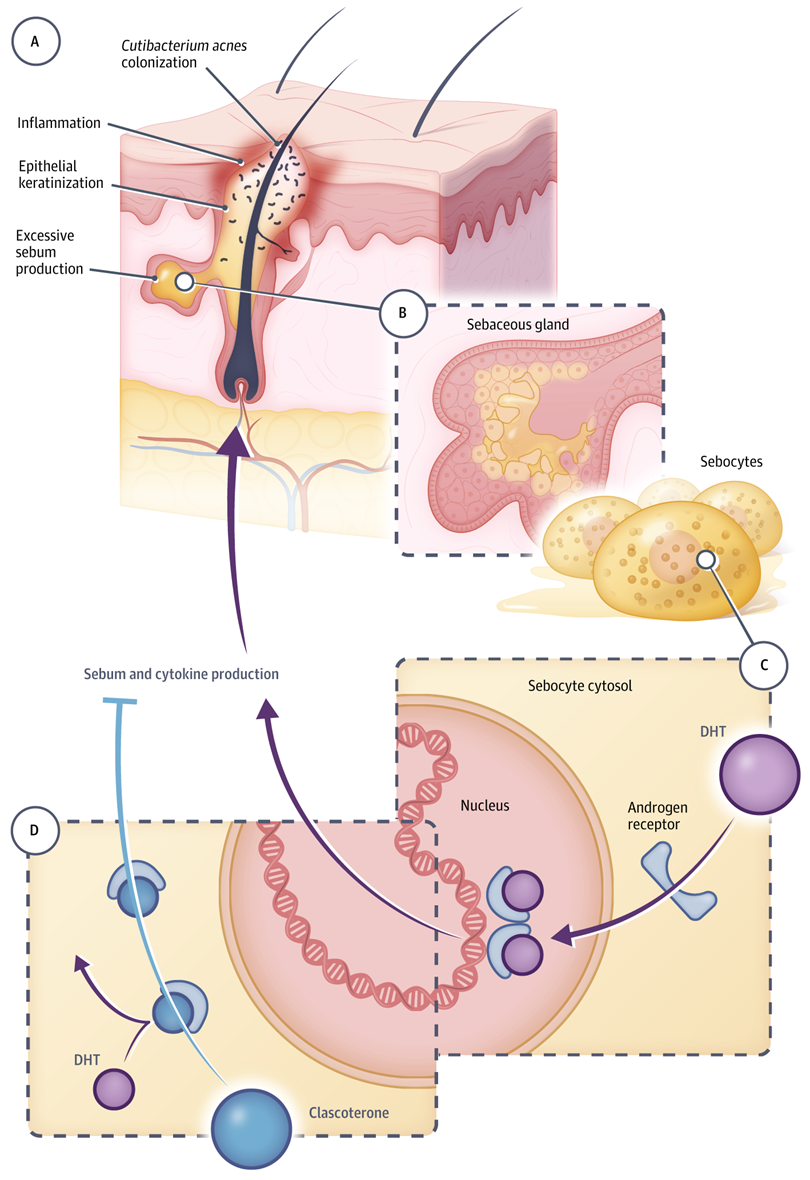
Cassiopea公司Winlevi:FDA批准40年来首款痤疮新机制疗法
2020年8月26日,美国FDA宣布批准
Cassiopea
公司开发的Winlevi(clascoterone)上市,用于治疗12岁及以上患者的痤疮(acne)。
痤疮是一种由于多种因素导致的皮肤疾病,受四种不同途径的影响:皮脂过多、毛孔阻塞(角化过度)、细菌生长和炎症。目前治疗痤疮的主要手段包括外用维A酸(retinoid),过氧化苯甲酰(benzoyl peroxide)和局部抗生素。虽然这些疗法对很多患者有效,但是仍然有不少患者的皮肤症状无法得到清除。而且,维A酸和过氧化苯甲酰都具有刺激性,导致有些患者难以使用这些疗法。激素在痤疮的病理发生中起到重要的作用。雄激素和其它促进油脂分泌的激素(例如IGF-1),能够导致皮脂分泌过多,过多的皮脂会促进痤疮丙酸杆菌的生长,皮脂成份的改变会激发角化过度和炎症。口服避孕药和螺内酯虽然能够有效针对痤疮的激素病理发生,但是它们可能导致潜在不良全身性反应,而且不能被男性和怀孕痤疮患者使用。
Winlevi是近40年来FDA批准的第一种具有新作用机制(MOA)的痤疮药物,是一种首创的(first-in-class)外用雄激素受体抑制剂,旨在解决男性和女性痤疮的雄激素成分。与治疗痤疮的口服激素不同,clascoterone 1%乳膏剂可以同时用于男性和女性患者。clascoterone是一种小分子药物,能够穿透皮肤到达皮脂腺和毛囊内的雄激素受体,该药是第一款没有系统性副作用的、安全有效的局部雄激素抑制剂疗法。临床结果显示,clascoterone 1%乳膏剂在所有主要临床终点表现出高度统计学意义的显著改善,证明了成功治疗痤疮和减少痤疮病变,并且一天2次使用时耐受性良好。
诺和诺德成人生长激素缺乏症新药Sogroya获FDA批准
2020年8月28日,美国FDA宣布批准诺和诺德(Novo Nordisk)公司开发的每周一次长效生长激素衍生物Sogroya(somapacitan-beco)上市,用于成人治疗生长激素缺乏症(GHD)。
GHD是一种以脑垂体前叶分泌生长激素不足为特征的疾病,垂体前叶是一个位于大脑底部的小腺体,可产生多种激素。生长激素是一种由脑垂体产生的蛋白质,调节生长和新陈代谢。GHD成人患者可接受生长激素作为一种替代疗法。
在治疗成人GHD方面,Sogroya是第一个每周只需皮下注射一次的人生长激素(hGH)疗法,而其他经FDA批准的hGH制剂必须每天注射。Sogroya的活性药物成分为somapacitan,这是一种hGH的长效类似物,采用了已应用于延长胰岛素、GLP-1半衰期方面将近20年的蛋白质技术。omapacitan由天然hGH经过修饰以增强其与血浆蛋白白蛋白(albumin)的结合,使其适合每周一次给药。目前,somapacitan正开发用于治疗成人和儿童的GHD,相关临床结果的评估由躯干脂肪的百分比变化确定,躯干脂肪是积聚在身体躯干或中心区域的脂肪,由生长激素调节。临床结果显示,在34周治疗期结束时,接受每周一次Sogroya组患者躯干脂肪平均减少了1.06%,而安慰剂组患者躯干脂肪增加了0.47%、每日一次Norditropin(somatropin,一种FDA批准的生长激素产品,诺和诺德公司产品)组患者躯干脂肪减少2.23%。每周一次Sogroya组患者和每日一次Norditropin组患者在其他临床终点也有类似的改善。
FDA批准放射性诊断剂Detectnet检测成人生长抑素受体阳性的神经内分泌瘤
2020年9月3日,美国FDA宣布批准由RadioMedix和Curium联合开发的放射性诊断试剂Detectnet(Cu 64 DOTATATE)上市,用于使用正电子发射断层扫描(PET),在成人中发现生长抑素(somatostatin, SST)受体阳性的神经内分泌瘤(NETs)。
NETs是一组起源于神经内分泌细胞的相对罕见的肿瘤。由于NETs细胞表面表达丰富的SST受体,放射性物质标记的SST类似物DOTATATE通过与SST受体结合,能够标记出肿瘤的位置。近年来已有多款不同放射性物质标记的DOTATATE获批,它们在肿瘤诊断,分期和治疗方面有着重要的地位。
Detectnet是首款将Cu 64放射性同位素与生长抑素类似物DOTATATE偶联的诊断试剂。临床结果显示,使用Cu 64 DOTATATE协助PET成像,在发现NETs方面的灵敏度达到90.9%,特异性达到96.6%。而且,与其它放射性元素偶联的DOTATATE相比,Cu 64 DOTATATE具有易于大规模生产,半衰期长的优点,有望让更多医师能够借助这一诊断试剂更及时地发现NETs。
Blueprint MedicinesRET非小细胞肺癌新药Gavreto获FDA批准
2020年9月4日,美国FDA宣布批准Blueprint Medicines公司开发的RET激酶抑制剂Gavreto(pralsetinib)上市,用于治疗经FDA批准的检测方法证实为RET融合阳性的转移性非小细胞肺癌(NSCLC)成人患者。
RET激活型融合和突变是许多癌症类型的关键疾病驱动因素,包括NSCLC和MTC。RET融合涉及约1-2%的NSCLC患者、约10-20%的甲状腺乳头状癌(PTC)患者,而RET突变牵涉到约90%的晚期MTC患者。此外,在结直肠癌、乳腺癌、胰腺癌和其他癌症中,也观察到低频率的RET改变,在耐药、EGFR突变的NSCLC患者中也观察到RET融合。
Gavreto是唯一一个每日口服一次的RET靶向疗法,在RET融合阳性NSCLC患者中显示出持久的疗效和高完全缓解率。其可选择性、强效抑制导致多种癌症的RET改变(融合和突变,包括预测的耐药突变),包括大约1-2%的NSCLC患者。临床结果显示:(1)在87例曾接受过含铂化疗的患者中, ORR为57%、CR为5.7%、mDoR尚未达到;(2)在27例未接受过含铂化疗的初治患者中,ORR为70%、CR为11%、,mDoR为9.0个月。
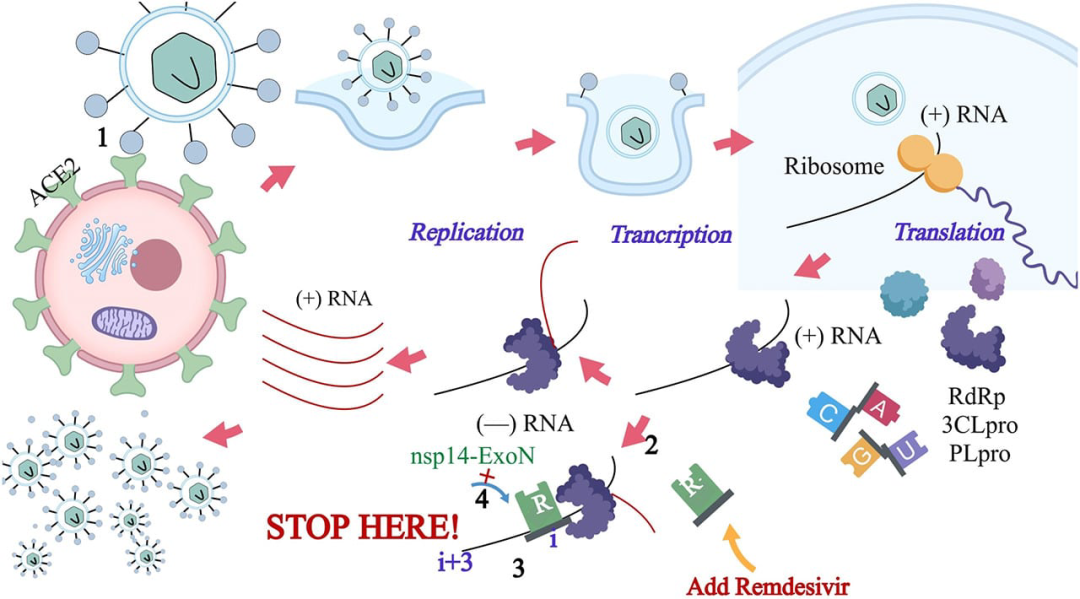
2020年10月22日,美国FDA宣布批准吉利德(Gilead)公司开发的Veklury(remdesivir)上市,用于治疗年龄在12岁及以上,体重至少40公斤,住院的COVID-19患者。
COVID-19肺炎以发热、干咳、乏力等为主要表现,少数患者伴有鼻塞、流涕、腹泻等上呼吸道和消化道症状。重症病例多在1周后出现呼吸困难,严重者快速进展为急性呼吸窘迫综合征、脓毒症休克、难以纠正的代谢性酸中毒和出凝血功能障碍及多器官功能衰竭等。值得注意的是重症、危重症患者病程中可为中低热,甚至无明显发热。轻型患者仅表现为低热、轻微乏力等,无肺炎表现。从目前收治的病例情况看,多数患者预后良好,少数患者病情危重。老年人和有慢性基础疾病者预后较差。儿童病例症状相对较轻。
Veklury是首个获FDA批准治疗COVID-19的药物。临床结果显示,与安慰剂和标准治疗相比,Veklury联合标准治疗在总体研究人群中加快了5天的恢复时间,在基线需要氧气支持的患者中加快了7天。Veklury还减缓了疾病的进展,与安慰剂相比,与 "死亡率降低的趋势 "有关。
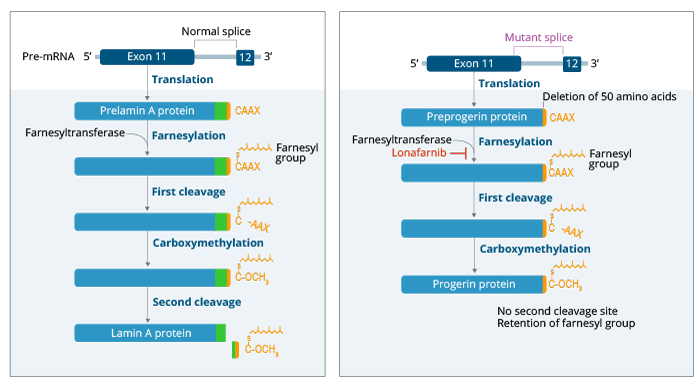
Eiger公司Zokinvy美国获批治疗早衰综合征
2020年11月20日,美国FDA宣布批准Eiger公司开发的口服法尼基转移酶抑制剂Zokinvy(lonafarnib)上市,用于减少哈金森-吉尔福德早衰综合征(HGPS)患者的死亡风险,以及治疗患有特定早老样核纤层蛋白病的1-2岁的患者。
HGPS又称为早年衰老综合症,儿童早老症,早衰症,是儿童罕见的加速衰老的致命遗传病。HGPS是人体内一种名叫LMNA(Lamin A)的蛋白基因发生突变导致。Lamin A基因错误编码导致支持核膜的结构蛋白质Lamin A少五十个氨基酸,蛋白progerin是lamin A蛋白的错误版本,通常情况下,lamin A帮助巩固细胞核,但是progerin却会导致畸形的细胞核和高于正常水平的DNA损伤。progerin可能破坏了干细胞替换损伤和死亡细胞的能力,从而促进了老化。
Zokinvy是美国FDA批准的首个早衰症疗法,其可以参与蛋白异戊二烯化修饰过程。通过抑制对早衰蛋白的异戊二烯化,它可以降低早衰蛋白在细胞核中的积累。临床结果显示,与哈金森-吉尔福德早衰综合征患者的历史数据相比,接受lonafarnib治疗的患者平均寿命分别延长3个月(治疗前3年)和2.5年(最长随访日期达11年),死亡率降低60%。
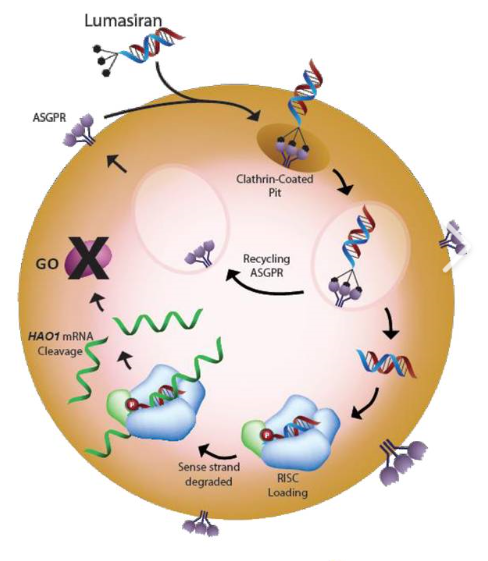
FDA批准首个治疗罕见代谢紊乱的药物:Alnylam制药Oxlumo上市
2020年11月23日,美国FDA宣布批准Alnylam制药公司开发的RNAi疗法Oxlumo(lumasiran)上市,用于治疗所有年龄段的原发性高草酸尿症1型(PH1)患者。
原发性高草尿酸症1型(PH1)是一种进行性、毁灭性的疾病,由过量的草酸生成导致肾衰竭,具有显著的发病率和死亡率,肝移植是目前唯一解决疾病根源的治疗方法。该病是一种非常罕见的、危及生命的疾病,影响肾脏和其他重要器官,该病影响婴儿、儿童和成人,患者面临着反复和痛苦的结石事件,以及肾功能进行性和不可预测的下降,最终导致终末期肾病,需要进行强化透析,作为肝/肾双重移植的桥梁。PH1通常在儿童期发病,需要立即进行有效的干预,晚期患者除了透析之外别无选择。
Oxlumo是首个被批准用于治疗PH1的药物,也是唯一一个被证明可以降低有害草酸水平的疗法。Oxlumo是一种靶向羟基酸氧化酶1(HAO1)的皮下注射RNAi药物, HAO1编码乙醇酸氧化酶(GO)。通过沉默HAO1和消耗GO酶,Oxlumo可抑制肝脏中草酸(直接参与PH1病理生理学的代谢物)的产生并使其正常化,从而潜在地阻止PH1疾病的进展。临床结果显示,与安慰剂组相比,Oxlumo治疗组尿草酸水平平均降低53%(p<0.0001)、与基线相比平均降低65%。此外,Oxlumo治疗组有84%的患者尿草酸水平达到正常或接近正常水平,安慰剂组为0%。
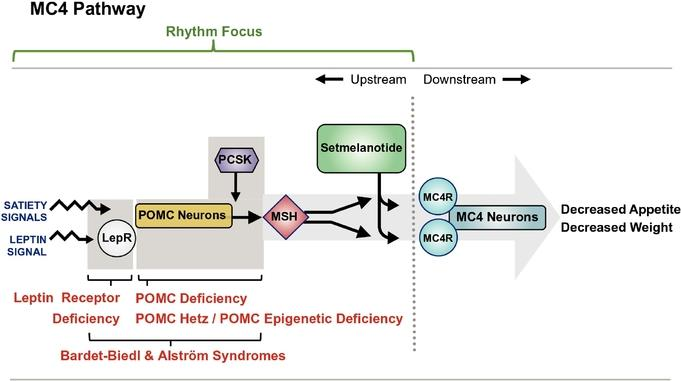
首个治疗特定基因缺陷引起的肥胖症新药,Rhythm制药Imcivree获FDA批准
2020年11月25日,美国FDA宣布批准Rhythm Pharmaceuticals,Inc.生物制药公司开发的Imcivree(setmelanotide)上市,用于因阿黑皮素原( POMC )、前蛋白转化酶枯草溶菌素1( PCSK1 )或瘦素受体( LEPR )基因缺陷导致肥胖的成人和儿童(6岁以上)患者的慢性体重管理。
POMC、PCSK1 或 LEPR 基因缺陷导致的肥胖是由于这些基因变异损害黑皮质素-4(MC4)受体通路,MC4受体通路是下丘脑中负责调节饥饿、能量消耗和体重的通路。由于 POMC 、 PCSK1 或 LEPR 基因缺陷而肥胖的患者,从年轻时就开始面临极端的、无法满足的饥饿感,导致早期发作的严重肥胖症。
Imcivree是FDA批准的首款用于治疗这些罕见遗传肥胖病的疗法。Imcivree 作为一款“first-in-class”MC4受体激动剂,旨在恢复由于MC4受体上游遗传缺陷而引起的受损的MC4受体通路活性。临床结果显示,Imcivree治疗一年后,有80%的因POMC或PCSK1缺乏症引起的肥胖症患者体重减轻超过10%,而45.5%的因LEPR缺乏症引起的肥胖症患者体重减轻超过10%。
放射性诊断剂Ga 68 PSMA-11美国获批诊疗前列腺癌
2020年12月1日,美国FDA宣布批准由加州大学洛杉矶分校和旧金山分校联合开发的镓 68 PSMA-11(Ga 68 PSMA-11)上市,用于疑似前列腺癌转移,并且通过手术或放射治疗可能治愈的患者。还适用于基于血清前列腺特异性抗原(PSA)水平升高而怀疑前列腺癌复发的患者。
Ga 68 PSMA-11是首款获批用于在前列腺癌男性患者中针对前列腺特异性膜抗原(PSMA)阳性病灶进行正电子发射断层扫描(PET)成像的药物。通过注射给药后,Ga 68 PSMA-11能够与PSMA结合,PSMA是前列腺癌成像的重要药理学靶点,在前列腺癌细胞通常高水平表达。作为一种发射正电子的放射性药物,Ga 68 PSMA-11可以通过PET成像,指示机体组织中存在的PSMA阳性前列腺癌病灶。临床结果显示,Ga 68 PSMA-11能够帮助确认癌症转移,并能检测出复发性前列腺癌患者的疾病部位,从而提供可能影响治疗方法的重要信息。
BioCryst公司Orladeyo美国获批预防遗传性血管性水肿发作
2020年12月3日,美国FDA宣布批准BioCryst公司开发的口服每日1次Orladeyo (berotralstat)上市,用于预防12岁及以上儿童和成人遗传性血管性水肿(HAE)发作。
HAE是一种罕见的遗传性疾病,据估计全世界每10万人中有2-10人患HAE。这种疾病会导致身体各个部位水肿,使人衰弱和疼痛,阻塞呼吸道的喉部水肿有可能危及生命。大多数HAE是由一种叫做C1酯酶抑制剂的蛋白质缺乏(1型HAE)或功能障碍(2型HAE)引起的。发作可以长达4天,可以是自发的,也可以是由压力、医疗程序和某些药物(如口服避孕药或ACE抑制剂)引发的,可能很少发作或者可能每隔几天发作一次,该疾病的严重性和不可预测性会显著降低患者的生活质量。
Orladeyo是血浆激肽释放酶抑制剂,与血浆激肽释放酶结合并抑制其蛋白水解活性。血浆激肽释放酶是一种蛋白酶,可将高分子量激肽原(HMWK)裂解为产生裂解的HMWK(cHMWK)和缓激肽,缓激肽是一种有效的血管扩张剂,可增加血管通透性,导致与HAE相关的肿胀和疼痛。在因C1而导致HAE的患者中,抑制剂(C1-INH)缺乏或功能障碍,血浆激肽释放酶活性不正常调节,这导致血浆激肽释放酶活性不受控制地增加,并导致血管性水肿攻击。Berotralstat会降低血浆激肽释放酶的活性,以控制HAE住院患者中过量的缓激肽生成。临床结果显示,完成48周治疗(150mg)的HAE患者中,HAE发作率在基线检查时为平均每月2.9次发作,治疗48周后降低至平均每月1.0次发作。在长期开放标签APeX-S试验中,完成48周治疗(150mg)的HAE患者,平均每月发作0.8次。
Athenex治疗光化性角化病新药Klisyri在美获批
2020年12月14日,美国FDA宣布批准Athenex和Almirall公司联合开发的Klisyri(tirbanibulin)上市,用于局部治疗面部或头皮的光化性角化病(actinic keratosis,AK)。光化性角化病是一种因为皮肤暴露在紫外线下导致的皮肤癌前病变,如果不及时治疗,10-15%的AK病变将发展为皮肤癌。
Klisyri(tirbanibulin)是一种微管抑制剂,通过抑制微管的聚合,它可以促进增生细胞发生细胞凋亡,适用于面部或头皮光化性角化病的局部治疗。临床结果显示,与赋形剂相比,在第57天,接受tirbanibulin治疗患者中的面部或头皮病灶完全清除的患者数量显著提高。
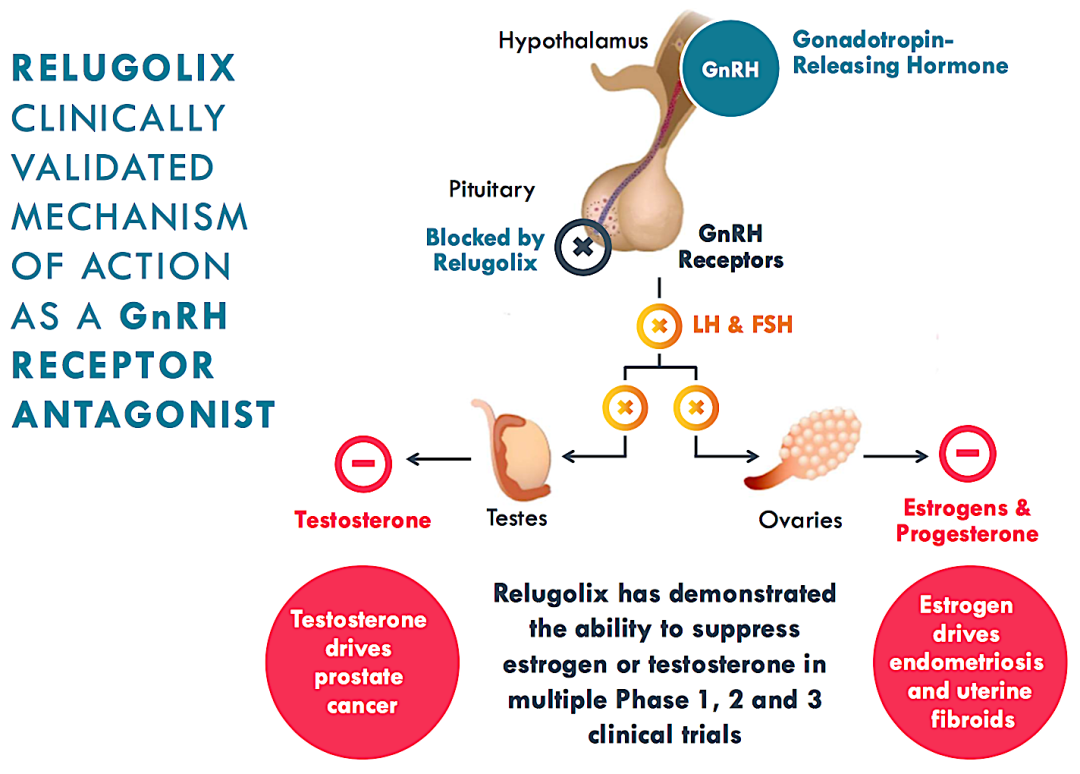
晚期前列腺癌首个口服药,武田Orgovyx获FDA批准
2020年12月18日,美国FDA宣布批准由武田、ASKA和Myovant共同开发的Orgovyx(Relugolix)上市,用于治疗晚期前列腺癌成人患者。
Orgovyx是美国FDA批准用于治疗晚期前列腺癌的第一个也是唯一一个口服促性腺激素释放激素(GnRH)受体拮抗剂。晚期前列腺癌的治疗方案之一是雄激素剥夺疗法(ADT),即通过药物降低帮助前列腺癌细胞生长的激素水平。目前,FDA批准治疗前列腺癌的ADT药物是注射剂或皮下植入剂,Orgovyx则是一种口服GnRH受体拮抗剂。Orgovyx能抑制睾丸睾酮的生成,这种激素可刺激前列腺癌细胞的生长。此外,relugolix也可通过阻断垂体腺中的GnRH受体,减少卵巢雌二醇的生成,这种激素已知可刺激子宫肌瘤和子宫内膜异位症的生长。
临床结果显示,Orgovyx治疗的缓解率高达96.7%,显著优于醋酸亮丙瑞林(为88.8%),同时将主要心血管不良事件(MACE)风险降低了54%。
Urovant Sciences膀胱过度活动症新药Gemtesa获FDA批准
2020年12月23日,美国FDA宣布批准Urovant Sciences公司开发的单克隆抗体Gemtesa(vibegron)上市,用于治疗伴急迫性尿失禁(UUI)、尿急、尿频的膀胱过度活动症(OAB)患者。
OAB是膀胱肌肉不自主收缩时发生的临床病症。症状可能包括尿急(难以控制的突然急迫性排尿)、急迫性尿失禁(在急迫性排尿后立即无意中排尿)、尿频(通常在24小时内排尿8次或更多次)和夜尿(夜间醒来排尿超过2次)。OAB令人烦恼的症状可能对患者的日常活动造成显著损害。
Gemtesa是第一种不需要剂量滴定的每日一次β3-激动剂,这种小分子β3肾上腺素能受体激动剂,有助于放松逼尿肌,使膀胱能容纳更多的尿液,从而减轻OAB症状。临床结果显示,与安慰剂组相比,Gemtesa治疗组患者每日UUI、排尿和急症发作次数显著减少,排尿量增加。
研发失败;不良副反应;中美临床数据桥接速度不及预期。
仅为公开资料整理,不构成投资建议。

使用本研究报告的风险提示及法律声明
兴业证券股份有限公司经中国证券监督管理委员会批准,已具备证券投资咨询业务资格。
本报告仅供兴业证券股份有限公司(以下简称“本公司”)的客户使用,本公司不会因接收人收到本报告而视其为客户。本报告中的信息、意见等均仅供客户参考,不构成所述证券买卖的出价或征价邀请或要约。该等信息、意见并未考虑到获取本报告人员的具体投资目的、财务状况以及特定需求,在任何时候均不构成对任何人的个人推荐。客户应当对本报告中的信息和意见进行独立评估,并应同时考量各自的投资目的、财务状况和特定需求,必要时就法律、商业、财务、税收等方面咨询专家的意见。对依据或者使用本报告所造成的一切后果,本公司及/或其关联人员均不承担任何法律责任。本报告所载资料的来源被认为是可靠的,但本公司不保证其准确性或完整性,也不保证所包含的信息和建议不会发生任何变更。本公司并不对使用本报告所包含的材料产生的任何直接或间接损失或与此相关的其他任何损失承担任何责任。
本报告所载的资料、意见及推测仅反映本公司于发布本报告当日的判断,本报告所指的证券或投资标的的价格、价值及投资收入可升可跌,过往表现不应作为日后的表现依据;在不同时期,本公司可发出与本报告所载资料、意见及推测不一致的报告;本公司不保证本报告所含信息保持在最新状态。同时,本公司对本报告所含信息可在不发出通知的情形下做出修改,投资者应当自行关注相应的更新或修改。
除非另行说明,本报告中所引用的关于业绩的数据代表过往表现。过往的业绩表现亦不应作为日后回报的预示。我们不承诺也不保证,任何所预示的回报会得以实现。分析中所做的回报预测可能是基于相应的假设。任何假设的变化可能会显著地影响所预测的回报。
本公司的销售人员、交易人员以及其他专业人士可能会依据不同假设和标准、采用不同的分析方法而口头或书面发表与本报告意见及建议不一致的市场评论和/或交易观点。本公司没有将此意见及建议向报告所有接收者进行更新的义务。本公司的资产管理部门、自营部门以及其他投资业务部门可能独立做出与本报告中的意见或建议不一致的投资决策。
本报告的版权归本公司所有。本公司对本报告保留一切权利。除非另有书面显示,否则本报告中的所有材料的版权均属本公司。未经本公司事先书面授权,本报告的任何部分均不得以任何方式制作任何形式的拷贝、复印件或复制品,或再次分发给任何其他人,或以任何侵犯本公司版权的其他方式使用。未经授权的转载,本公司不承担任何转载责任。
在法律许可的情况下,兴业证券股份有限公司可能会持有本报告中提及公司所发行的证券头寸并进行交易,也可能为这些公司提供或争取提供投资银行业务服务。因此,投资者应当考虑到兴业证券股份有限公司及/或其相关人员可能存在影响本报告观点客观性的潜在利益冲突。投资者请勿将本报告视为投资或其他决定的唯一信赖依据。
投资评级说明
报告中投资建议所涉及的评级分为股票评级和行业评级(另有说明的除外)。评级标准为报告发布日后的12个月公司股价(或行业指数)相对同期相关证券市场代表性指数的涨跌幅,A股市场以上证综指或深圳成指为基准。 行业评级:推荐-相对表现优于同期相关证券市场代表性指数;中性-相对表现与同期相关证券市场代表性指数持平;回避-相对表现弱于同期相关证券市场代表性指数。 股票评级:买入-相对同期相关证券市场代表性指数涨幅大于15%;审慎增持-相对同期相关证券市场代表性指数涨幅在5%~15%之间;中性-相对同期相关证券市场代表性指数涨幅在-5%~5%之间;减持-相对同期相关证券市场代表性指数涨幅小于-5%;无评级-由于我们无法获取必要的资料,或者公司面临无法预见结果的重大不确定性事件,或者其他原因,致使我们无法给出明确的投资评级。
免责声明
市场有风险,投资需谨慎。本平台所载内容和意见仅供参考,不构成对任何人的投资建议(专家、嘉宾或其他兴业证券股份有限公司以外的人士的演讲、交流或会议纪要等仅代表其本人或其所在机构之观点),亦不构成任何保证,接收人不应单纯依靠本资料的信息而取代自身的独立判断,应自主做出投资决策并自行承担风险。根据《证券期货投资者适当性管理办法》,本平台内容仅供兴业证券股份有限公司客户中的专业投资者使用,若您并非专业投资者,为保证服务质量、控制投资风险,请勿订阅或转载本平台中的信息,本资料难以设置访问权限,若给您造成不便,还请见谅。在任何情况下,作者及作者所在团队、兴业证券股份有限公司不对任何人因使用本平台中的任何内容所引致的任何损失负任何责任。
本平台旨在沟通研究信息,交流研究经验,不是兴业证券股份有限公司研究报告的发布平台,所发布观点不代表兴业证券股份有限公司观点。任何完整的研究观点应以兴业证券股份有限公司正式发布的报告为准。本平台所载内容仅反映作者于发出完整报告当日或发布本平台内容当日的判断,可随时更改且不予通告。
本平台所载内容不构成对具体证券在具体价位、具体时点、具体市场表现的判断或投资建议,不能够等同于指导具体投资的操作性意见。



 个人中心
个人中心
 我是园区
我是园区
 退出
退出