

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!
AYVAKIT(AVAPRITINIB)
1月9日,FDA批准了Blueprint Medicines公司的avapritinib,用于阿尔法血小板衍生生长因子受体(PDGFRA)基因外显子18突变阳性,且无法手术切除或转移性的成人胃肠间质瘤患者治疗。胃肠间质瘤是一种比较罕见的肿瘤,美国每年新发病例为5000-6000人[i],其中大约有10%左右的病例携带PDGFRA基因突变。虽然FDA之前已经批准了多个用于胃肠间质瘤的药物,但PDGFRA基因突变的患者对标准疗法不应答,avapritinib是首个获批用于PDGFRA基因突变阳性胃肠间质瘤的疗法。此次FDA批准avapritinib,是基于一项有43名患者参与的临床研究(NCT02508532)结果。入组的43名患者全部为外显子18突变,其中38名患者为PDGFRA D842V突变。入组患者接受每日一次300mg或400mg的avapritinib治疗,直至疾病进展或出现不可接受的毒性,结果显示总缓解率为84%,其中7%完全缓解,77%部分缓解,而PDGFRA D842V突变的患者亚组中,总缓解率达89%,8%的患者完全缓解,82%的患者部分缓解。虽然尚未达到中位持续缓解时间,但61%的患者持续缓解时间超过了6个月,而PDGFRA D842V突变的亚组中,这一数字也达到了59%[ii]。不良反应方面,根据一项1期临床试验的结果显示,与治疗相关的3-4级不良反应发生率为57%,其中最常见的不良反应为贫血(17%)[iii]。市场潜力方面,虽然BlueprintMedicines还在开发胃肠间质瘤以外的适应症,但结合avapritinib在美国的定价和适用患者数量估算,该产品在未来五年内的年销售额突破1亿美元具有较大的难度。
TEPEZZA(TEPROTUMUMAB-TRBW)
1月21日,FDA批准了HORIZON THERAPEUTICS的teprotumumab,用于甲状腺眼病的治疗。甲状腺眼病又称Graves眼病,是一种自免疫相关的疾病,年发病率为19/10万[iv],然而这种疾病在美国患病相对较低,目前尚无准确的流行病学数据,根据美国罕见疾病组织(NORD)数据,该疾病的女性患病率为16/10万,男性患病率为2.9/10万[v]。FDA批准teprotumumab上市,是基于两项、共有170名患者参与的临床试验的结果,其中teprotumumab治疗组分别有71%(试验一,NCT01868997)和83%(试验二,NCT03298867)的患者眼球突出高度下降了2mm以上,而安慰剂组患者则分别只有20%和10%达到这一标准。Teprotumumab是首个专门获批用于甲状腺眼病的药物,对于甲状腺眼病的治疗是一座重要的里程碑[vi]。市场方面,虽然甲状腺眼病被FDA定义为罕见病,但患者基数非常可观,在“孤儿药”的高价之下,本品将成为HORIZON的“现金牛”,根据该公司三季度财报,该产品在过去两个季度的销售额就已达4.76亿美元,并且预测2020年的年销售额将超过8亿美元,美国的销售额峰值超过30亿美元[vii]。
TAZVERIK(TAZEMETOSTAT)
1月23日,FDA加速批准了EPIZYME 公司的tazemetostat用于转移性、局部转移性或无法手术根除的晚期上皮样肉瘤治疗。上皮样肉瘤是一种非常罕见软组织肿瘤,仅占所有软组织肉瘤的比例不到百分之一,初步估算美国仅有数百名患者。在此之前,上皮样肉瘤没有特效的治疗方法,tazemetostat是一种甲基转移酶抑制剂,可阻断EZH2甲基转移酶的活性,进而阻止癌细胞的生长。FDA加速批准本品上市,是基于一项有62名转移性或局部晚期上皮样肉瘤患者参与的开放标签的临床研究(NCT02601950)结果。入组患者接受每日两次800mg的tazemetostat治疗,直到疾病进展或患者出现不可耐受的毒性为止。结果显示,总缓解率为15%,其中1.6的患者完全缓解,13%的患者部分缓解,9名患者中,6名患者的持续缓解时间超过了6个月[viii]。除了上皮样肉瘤,tazemetostat还有多个在研的适应症,其中走在最前沿的是滤泡性淋巴瘤。在一项2期临床试验中,45名EZH2基因突变型患者和54名EZH2基因野生型患者,分别接受本品治疗,结果显示两组患者的客观缓解率分别为69%和35%,应答患者所获得的中位持续缓解时间分别为10.9个月和13.0个月,中位无进展生存期为13.8个月和11.1个月[ix]。市场方面,由于本品适用的患者非常少,年销售额很难突破10亿美元。
PIZENSY(LACTITOL)
2月12日,FDA批准了BRAINTREELABS的乳糖醇(lactitol),用于成人慢性特发性便秘(CIC)治疗。乳糖醇是一种渗透型泻剂,FDA批准本品上市是基于两项临床试验的结果。在其中一项随机、双盲的临床试验(NCT02819297)中,入组患者分别基于乳糖醇和安慰剂治疗6个月,以每周自发排便数超过3次、12周内至少有9周且最后4周至少有3周的自发排便数相比基线至少增加1次为综合指标,计算患者的应答率,结果显示291名使用乳糖醇治疗的患者获得的应答率为25%,而使用303名使用安慰剂治疗的患者的应答率仅为13%,达统计学差异。而另外一项临床试验(NCT02481947)则是以鲁比前列酮为对照,评估方法与前一项临床试验一致,乳糖醇所获得的治疗结果也与前一项临床试验结果相似[x],但非劣结果未被FDA采纳。乳糖醇是老药新批的又一典范,本品早在1993年就在西班牙获批上市,除了便秘,西班牙还批准乳糖醇用于肝性脑病治疗[xi]。
NEXLETOL(BEMPEDOICACID)
2月21日,FDA批准了ESPERIONTHERAPS的bempedoic acid,用于在饮食控制和他汀类最大耐受剂量治疗的下,仍需要进一步降低脂蛋白(LDL-C)的杂合子高胆固醇血症或动脉粥样硬化心血管疾病患者治疗。Bempedoic acid是一种新机制的口服降脂药,可通过抑制三磷酸腺苷-柠檬酸裂解酶(ACL)的活性,降低肝脏对LDL-C的合成。FDA批准本品上市是基于两项随机、双盲的临床研究结果,在第一项临床试验(NCT02666664)中,2330名患者在接受最大耐受剂量降脂治疗(最大剂量的他汀或他汀类联用其它药物,但不含使用40mg及以上辛伐他汀或PCSK9抑制剂的患者)的基础上,按2:1的比例分别添加本品或安慰剂治疗,结果显示本品治疗组在第12周的LDL-C水平相比基线下降17%,而安慰剂组仅为2%,达统计学差异[xii]。另一项临床试验(NCT02991118)的设计和结果与前一项试验相似,bempedoic acid和安慰剂治疗组在第12周的LDL-C水平分别相比基线下降15%和2%[xiii]。市场方面,虽然本品面临着PCSK9抑制剂的竞争,但美国有超过50%的患者对他汀类不耐受或单独使用他汀类无法有效控制血脂,因此ESPERION对bempedoic acid寄予厚望,认为Nexletol和Nexlizet(bempedoic acid+依折麦布)可拿下20亿美元的年销售额。
VYEPTI(EPTINEZUMAB)

BARHEMSYS(AMISULPRIDE)
2月26日,FDA批准了ACACIA公司的氨磺必利注射液, 用于术后呕吐预防和治疗。氨磺必利是一个多巴胺D2和D3受体阻断剂,上世纪80年代开始应用于精神分裂症治疗。Barhemsys将是老药新用史上的又一大成功典范,在此之前,FDA批准用于术后呕吐治疗预防的药物主要是五羟色胺受体3阻断剂和地塞米松,本品的获批将为患者带来全新的治疗选择,尤其是其它止吐药疗效不理想的患者。FDA此次批准氨磺必利用于术后呕吐治疗和预防的依据是4项临床研究的结果,其中试验一和实验二是以预防为设计目的的临床试验,而试验三和试验四则是以治疗为设计目的,试验一(NCT01991860)[xviii]入组的患者均接受本品或安慰剂单独预防(n=342),试验二(NCT02337062)[xix]入组的患者均接受本品或安慰剂联合其它机制的止吐药共同预防(n=1147),试验三(NCT02449291)[xx]入组的患者均未接受过任何止吐药治疗(n=369),试验四((NCT02646566)[xxi]入组的患者均为既往接受过其它止吐药治疗(n=465),临床终点均为完全缓解(试验一、二定义为术后24小时内无呕吐发作或不使用急救药物,试验三、四定义为使用药物治疗后24小时内无呕吐发作或不使用急救药物)率,结果显示,4项临床试验的完全缓解率依次分别为(治疗组 vs 安慰剂组):78% vs 54%、58% vs 47%,31% vs 22%和42% vs 29%,均达到统计学差异。
NURTEC ODT(RIMEGEPANT)
2月27日,FDA批准了Biohaven的口服CGRP抑制剂rimegepant,用于成人偏头痛的急性治疗。Rimegepant不同于CGRP单抗,该产品的定位是快速止痛,为了加快药物的起效速度,Biohaven将其制成了口崩片。FDA批准本品上市是基于一项随机双盲的临床试验((NCT03461757),入组患者分别接受本品75mg(n=732)和安慰剂(n=734)治疗,试验的主要终点为单次服药后2小时内无痛患者应答率和无自主制定的最明显症状(MBS,主要是畏光、恶心、恐声)患者应答率。结果显示,本品治疗组有21.2%的无痛应答和35.1%的无最明显症状应答,而安慰剂组分别仅为10.9%和26.8%,均达到统计学差异[xxii]。另一项相似设计、有1186名患者参与的多中心临床试验(NCT03237845)也证实了本品的有效性,其结果显示本品治疗组有19.6%的无痛应答和37.6%的无最明显症状应答,而安慰剂组分别仅为12.0%和25.2%,也均达到了统计学差异[xxiii]。市场方面,因为本品作为第一个口服CGRP抑制剂,而且市场定位与注射的CGRP单抗不同,可以在偏头痛市场中获得一席之地,根据Biohaven的3季度财报,该公司仅在3季度就已经卖出1770万美元,如果预防性治疗偏头痛sNDA(补充)申请获得FDA批准,该产品的年销售额突破一亿美元并非难事,销售额峰值甚至可超过10亿美元。除了对欧美市场寄予厚望,Biohaven也非常重视中国市场,rimegepant是否能打开中国偏头痛的市场,非常值得期待。
SARCLISA (ISATUXIMAB-IRFC)
3月2日,FDA批准了赛诺菲的ISATUXIMAB,联合泊马度胺和地塞米松用于既往接受过两种以上疗法(包括泊马度胺和蛋白酶体抑制剂)的多发性骨髓瘤(MM)治疗。Isatuximab 是一种Anti-CD38抗体,通过抑制其ADP核糖环化酶(ADP-ribosyl cyclase)活性而发挥生理作用。一项名为ICARIA-MM (NCT02990338)的临床试验评估了本品与泊马度胺和地塞米松的安全有效性。307名符合入组标准的多发性骨髓瘤患者按1:1的比例随机分组,分别在泊马度胺和地塞米松治疗的基础上,增加本品或安慰剂治疗,结果显示本品治疗组患者客观缓解率为60.4%,中位无进展生存期(PFS)为11.53个月,而安慰剂组则分别仅为35.3%和6.47个月,均达到统计学差异,中位总生存期(OS)尚未达到[xxiv]。多发性骨髓瘤是比较少见的一种肿瘤,美国美国国家癌症研究所数据显示,美国2020年将有32270名新发病例和12830名死亡病例[xxv],故isatuximab顺利获得了FDA孤儿药认定。尽管如此,在isatuximab获批之前,FDA已经批准了强生的另一个CD38抗体daratumumab,相比isatuximab,daratumumab获准的应用范围更加广泛,而且目前已经可以实现皮下注射治疗,因此本品仅凭MM一个适应症就想成为年销售额超过10亿美元的“重磅炸弹”难度较大,好在isatuximab用于多种淋巴瘤和白血病治疗的临床试验正在有序开展,如果进展顺利,未来潜力同样值得关注。
ISTURISA(OSILODROSTAT)
3月6日,FDA批准了RECORDATI公司的OSILODROSTAT片,用于不适宜垂体手术或经过手术未治愈的成年库欣病患者治疗。库欣病是由垂体瘤引起,垂体释放出过多促肾上腺皮质激素引起肾上腺皮质醇合成过量,进而引发一些列的临床症状,如高血压,肥胖,2型糖尿病,骨质疏松等等。库欣病是一种非常罕见的疾病,美国国家罕见病组织数据显示,该病的年发病率仅百万分之十三[xxvi]。Osilodrostat是一种皮质醇合成抑制剂,可抑制11β-羟化酶的活性,阻断肾上腺生物合成皮质醇。FDA批准本品上市是基于一项有137名患者参与的临床试验,入组患者均不宜接受或不接受垂体手术治疗,经过24周的开放标签治疗,约有一半患者的皮质激素水平下降至正常范围内。在此基础上,71名无需增加治疗剂量(30mg每日2次)且在过去12周内耐受该药物的患者被纳入为期8周的双盲、随机停药研究,结果显示,86%继续使用osilodrostat治疗的患者皮质激素维持在正常水平,而停药使用安慰剂治疗的患者仅为30%[xxvii]。
ZEPOSIA(OZANIMOD)
3月25日,FDA批准了CELGENE的ozanimod,用于治疗成人复发型多发性硬化症(RMS),包括临床孤立综合征、复发缓解性疾病、活动性继发进展性疾病。Ozanimod是一种鞘氨醇1-磷酸(s1p)受体调节剂,与s1p 1和5有非常高的亲和力,但目前该产品在多发性硬化中的作用机制尚不完全清楚。FDA批准本品上市是基于两项随机、双盲、安慰剂对照的临床试验的结果。试验一((NCT02294058)共计有895名患者入组,持续治疗1年,试验二(NCT02047734)共有874名患者入组,持续治疗2年,入组患者均接受本品和β干扰素1a治疗,主要终点均为治疗期内的年化复发率。试验一结果显示,本品治疗组的年化复发率为0.181,78%的患者未出现复发,β干扰素治疗组年化复发率为0.350,66%的患者未出现复发,达治疗终点[xxviii];试验二结果显示本品治疗组的年化复发率为0.172,76%的患者未出现复发,β干扰素治疗组年化复发率为0.276,64%的患者未出现复发,也达到了治疗终点[xxix]。除了MS,ozanimod正在开发的适应症包括克罗恩病和炎性肠病,这两个适应症如能开发成功,ozanimod将可能是不折不扣的“重磅级炸弹”。
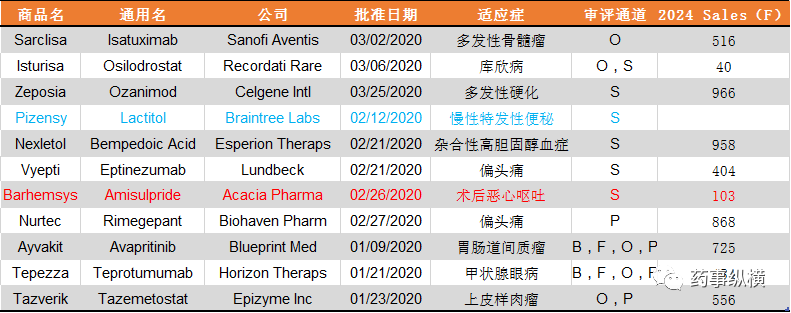
注:表中O代表孤儿药,B代表突破性疗法,P代表优先审评,F代表快速通道,S代表标准审评,销售额预测来自Evaluatepharma,原始销售额预测数据来源于以下三个公开网址:
https://www.evaluate.com/vantage/articles/news/us-fda-approval-tracker-first-aimmune
https://www.evaluate.com/vantage/articles/news/snippets/us-fda-approval-tracker-february
https://www.evaluate.com/vantage/articles/news/snippets/us-fda-approval-tracker-march
本文内容为药事纵横原创,每一个数据都有确切的来源,但受篇幅影响,参考文献的部分已省略,未经允许不得私自转载、抄袭,侵权必究。

药事纵横是一个开放,由自愿者组成的团体,由以下成员组成:Voyager88,雷道安,Herman,梅希,文竹,duke,子炎,ZMJ和曾文亮。欢迎有志之士加入我们团队。投稿、合作、加专业群请加微信437180999,药事纵横二千人QQ群22711855






