Nat Chem Biol | 何蓉蓉团队等报道铁死亡新调控蛋白iPLA2β——逆转铁死亡或为治疗帕金森病的新策略
收藏
关键词:
治疗Nat新策略
资讯来源:BioArt + 订阅账号
所属行业:化学药制剂 + 订阅行业
发布时间:
2021-02-05
多巴胺能神经元退行性死亡是临床上导致PD的主要病理机制,目前研究认为凋亡、坏死性凋亡和
自噬性死亡等方式都参与了多巴胺能神经元的退行性丢失过程。
但是,这些死亡方式都不足以解释其病理进程和机制,深入研究发现这些死亡方式都存在一个共同特征,即:
多巴胺能神经元细胞的脂质过氧化损伤。
真正能反应细胞脂质过氧化的细胞死亡方式是
近些年新发现的细胞死亡方式——铁死亡。
铁死亡主要特征是以铁离子的负荷增多为驱动因素和大量脂质过氧化物为细胞致死因素,而这些特征与临床上帕金森病人的脑变化分子生物学特征高度吻合。
暨南大学何蓉蓉教授团队长期关注情志应激诱导的细胞氧化应激损伤对帕金森病“易感性”影响,并提出多巴胺能神经元磷脂氧化可能是帕金森病的重要病理机制。
Kagan教授课题组之前发现,15-LOX在伴侣分子PEBP1的作用下能够锚定到细胞膜上,特异性氧化磷脂酰乙醇胺
(PE)
生成氧化型磷脂酰乙醇胺
(ox-PE)
,其中PE氧化产物15-HpETE-PE是细胞发生铁死亡的重要信号分子
(Nat Chem Biol. 2017, 13:81-90;Cell. 2017, 171:628-641)
。
双方在合作过程中证实多巴胺能神经元磷脂氧化参与了帕金森的病理过程,且其机制与氧化磷脂的修复不力有关。
生物膜内磷脂的不饱和脂肪酸链不断被氧化,又不断被修复,如果修复不及时导致磷脂过氧化物大量堆积就可能引发细胞发生铁死亡。在氧化磷脂修复过程中,
钙非依赖磷脂酶A2β
(
iPLA2β
,对应基因名称是PNPLA9)发挥了重要作用。iPLA2β是一种特异性水解磷脂的sn-2酰基键的蛋白,对细胞膜氧化磷脂清除和磷脂的重构具有重要作用。临床研究显示,
PNPLA9突变和帕金森病发病密切相关,但是其发病机制却不清楚。临床中已经发现80多种不同的PNPLA9突变类型,在该研究中作者重点关注PNPLA9R747W突变类型的发病机制,所获结论对其他突变类型也适用。
近日,暨南大学
何蓉蓉教授团队和匹兹堡大学
Valerain E. Kagan教授团队合作在
Nature Chemical Biology上在线发表了题为
“Phospholipase iPLA2β Averts Ferroptosis By Eliminating A Redox Lipid Death Signal”的研究工作。该研究利用氧化脂质组学等技术阐明了磷脂重塑关键蛋白iPLA2β突变导致其水解氧化磷脂ox-PE活力下降,导致多巴胺能神经元中氧化磷脂堆积。
该研究发现iPLA2β是铁死亡的一个重要调节蛋白,其活性丧失引起的脂质过氧化物累积与帕金森病的发生紧密相关,抑制铁死亡有望成为治疗帕金森病的一种新策略。
在这项研究中,研究人员从临床现象
(iPLA2β突变帕金森病患者细胞存在明显氧化磷脂堆积现象)出发,采用健康人细胞H109和有iPLA2β突变的帕金森病患者细胞fPDR747W,提取细胞磷脂进行氧化磷脂组学实验。实验结果显示,与正常人细胞H109相比,fPDR747W细胞的磷脂过氧化水平明显更高,铁死亡氧化磷脂信号分子15-HpETE-PE水平也明显更高。
进一步为了探讨iPLA2β缺陷和铁死亡的关系,作者分别给予H109和fPDR747W细胞RSL3,观察细胞活力变化。当iPLA2β功能缺失时,细胞对RSL3导致的铁死亡更加敏感。这一现象也在iPLA2β蛋白敲低和敲除细胞中被观察到,说明iPLA2β能够一定程度抑制细胞发生铁死亡。
何蓉蓉教授团队采用CRISPR-Cas9技术构建Pnpla9R748W点突变小鼠
(对应人R747W突变位点)验证iPLA2β功能和帕金森病发病的关系。与WT小鼠相比,纯合子突变小鼠自3月龄开始出现爬杆、转棒、步态行为异常
(与常用帕金森病SncaA53T基因小鼠模型相比,该模型发病时间明显缩短);7月龄纯合子突变小鼠中脑中多巴胺及其代谢物水平显著降低,4-HNE水平显著升高,GSH耗竭,并且伴随铁死亡信号分子15-HpETE-PE水平显著升高。体外酶活实验结果显示,突变小鼠中脑iPLA2β表达水平未改变,但是催化活力显著降低。
为探讨磷脂过氧化和帕金森病发病的关系,研究人员进一步建立了鱼藤酮诱导和SCNA-A53T的鼠源帕金森病模型。对鱼藤酮诱导大鼠的黑质进行氧化脂质组学分析,发现ox-PE显著升高。连续给药鱼藤酮10-14天时,大鼠黑质部位中15-HpETE-PE累积,并且iPLA2β活力显著降低。对8月龄SncaA53T小鼠模型进行研究,发现突变小鼠中脑中15-HpETE-PE累积,并且伴随iPLA2β表达量显著降低。上述结果说明
iPLA2β功能缺失引起的脂质过氧化物累积现象也存在于其它帕金森疾病模型中。
使用iPLA2β WT和R747W分别对非氧化磷脂1-SA-2-ETE-PE和氧化磷脂1-SA-2-15-HpETE-PE两种底物进行体外催化反应,发现iPLA2β对1-SA-2-15-HpETE-PE的催化活性高于1-SA-2-ETE-PE,且当iPLA2β点突变后,该酶对1-SA-2-15-HpETE-PE的催化活性大幅降低。类似的结果也在帕金森病患者突变皮肤纤维细胞fPDR747W中被观测到。
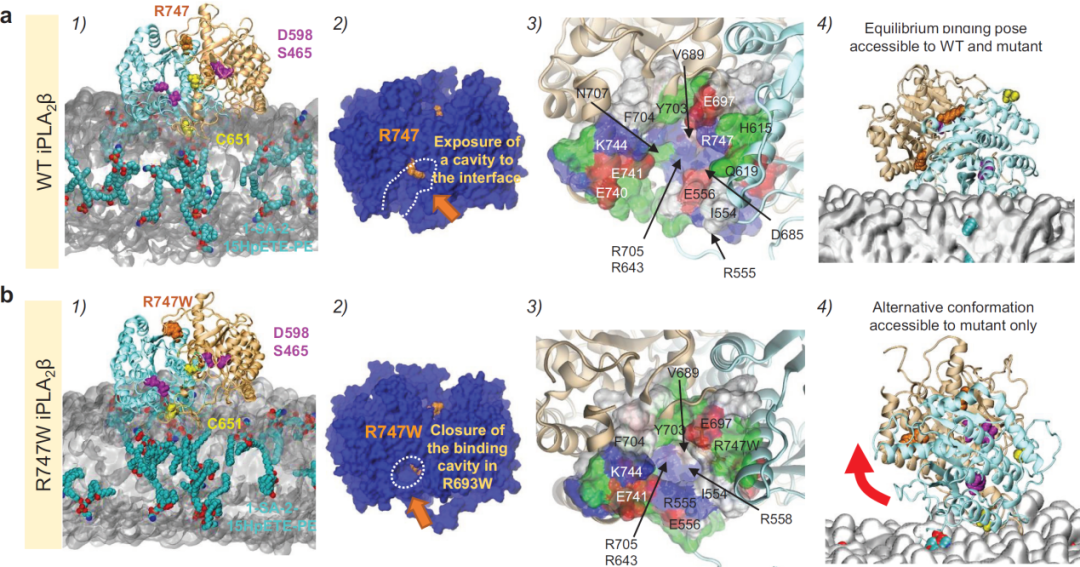
为理解突变导致iPLA2β功能缺失的机制,进一步采用计算机模拟iPLA2β WT和R747W二聚体与细胞膜磷脂的相互作用
(下图)。野生型蛋白锚定到细胞膜后,磷脂容易进入活性口袋,接近活性中心位点,有利于酶催化反应。蛋白突变后,活性口袋缩小,磷脂难以接近活性中心位点,使得酶催化反应受阻。此外,突变蛋白还产生了一个新的无催化活力的结合构象。
突变导致iPLA2β和细胞膜磷脂结合构象改变,酶解氧化磷脂的能力降低
本研究主要揭示了多巴胺能神经元磷脂氧化堆积是帕金森病的重要病理机制,证明了iPLA2β功能缺失能够导致多巴胺能神经元铁死亡“易感”,而且 iPLA2β抗铁死亡功能主要是清除铁死亡的死亡信号分子1-SA-2-15-HpETE-PE。
何蓉蓉团队骨干成员孙万阳
博士为第一作者,何蓉蓉教授、匹兹堡大学Valerian Kagan教授及其团队成员Yulia Tyurina
为共同通讯作者。
https://doi.org/10.1038/s41589-020-00734-x


 个人中心
个人中心
 我是园区
我是园区
 退出
退出