

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

•《专利法实施细则》及《专利审查指南》的征求意见稿将 “新药”的含义界定为“全球新”。
• 专利期补偿制度中参考药品法体系中已经废止的“新药申请”中的“全球新”概念,将新药的定义对标到药品审查中的一个具体程序上是否过窄?

过去几年5.1类进口药获批的数量远超1类创新药;同时,国内的License-in交易数量在攀升。不过,这些5.1类进口药以及License-in引入的产品,很可能将无法获得专利期补偿。
按照目前《专利法实施细则》和《专利审查指南》征求意见稿中“全球新”的概念,适用于药品专利期补偿制度的“新药”仅包括1类创新药和部分2类改良型新药,5.1类进口药以及License-in产品将被排除在外。
“从专利法的立法目的、专利法与药品法的关系以及国际上关于该制度的实践经验等角度来看,将‘新药’定义为‘全球新’能否真正实现鼓励创新的目的还值得商榷。”在研发客主办的主题为“当License-in模式遇到药品专利期限的补偿机制”的网络研讨会上,北京务实知识产权发展中心的程永顺主任表示。
《专利法实施细则》和《专利审查指南》正在征求意见,依据《立法法》规定的立法进度,实施细则预计将在今年5月底前正式出台。
“全球新”是否合理?

观点
按照《专利法》立法者此前的相关说明,引入药品专利期补偿制度的立法目的,旨在将其作为Bolar例外(Bolar例外条款规定了仿制药企业就可以在专利药品的专利到期之前,提前进行相应的生产和准备,提供行政审批所需的信息,从而在原研药品的专利到期之后能够立即进入市场)的配套制度,实现与仿制的平衡,并促进国外新药能够尽早在我国上市,提高药品可及性,保障公共健康。
然而,每一项制度规则的制定和出台都应有其自身的特定目的,不应指望该制度解决该领域的所有相关问题。想要通过药品专利期补偿制度,既要鼓励、保护药品创新,又要提升消费者用药福祉,提高可用药品多样性,这是这个制度无法承受之重,在目前的背景下也不一定能达到理想的效果。
提高人民用药可及性是药品法应当考虑的问题,不应直接作为专利保护期补偿制度的制度目标,更不宜将其作为专利法的立法目的,认为因为实施了药品专利期补偿制度,所以可以实现药品可及性的观点则混淆了药品专利期补偿制度与实现药品可及性之间的因果关系。这两者之间的因果关系应当是:因为药企选择在中国境内上市,而这种选择将有助于为中国患者提供更多的用药选择,因此,对于这些首次在中国上市的药品相关的专利予以专利期补偿,这是从制度层面对这些药企作出进入中国市场的商业选择和商业决策的鼓励措施。
另一方面,虽然药品专利期补偿制度同时涉及药品法和专利法,但实际上,这两部法律的性质、立法目的,所调整的法律关系存在较大差异,相关制度规则的设计和法律术语的界定和解释也应当在其自身的制度框架内进行。
药品法属于公法领域,要求监管部门加强对药品的管理,保证药品的安全性和有效性,对人民的健康负责;而专利法属于私法领域,处理的是作为私权的专利权的相关法律关系,其立法目的是保护专利权人的合法权益,鼓励和推动药物的发明创新。尽管立法者试图保持法律体系中对于相同法律术语的一致性解释,但目前,专利期补偿制度中参考药品法体系中已经废止的“新药申请”中的“全球新”概念,将新药的定义对标到药品审查中的一个具体程序上是否过窄是我们应该思考的问题。
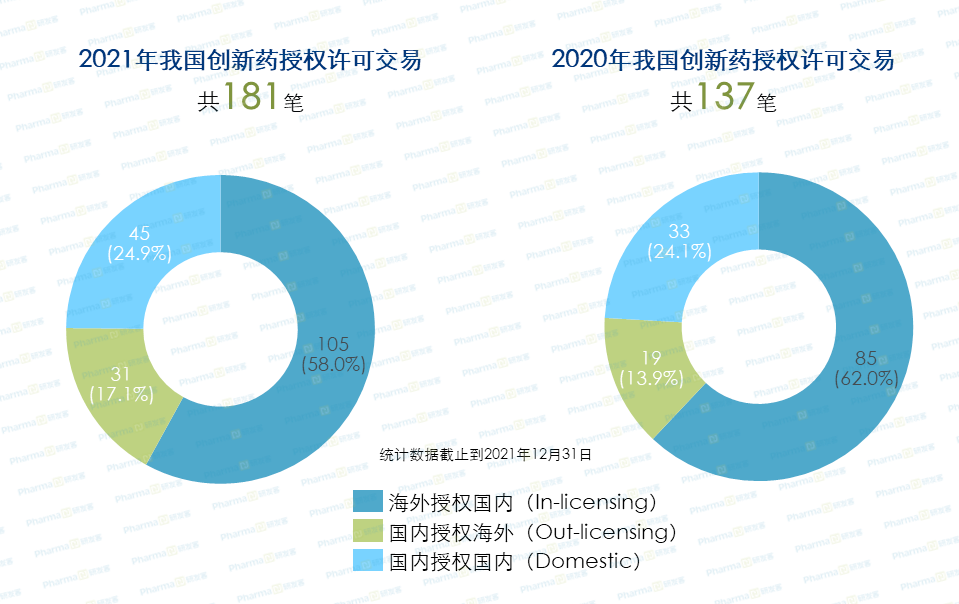
不容忽视的现实背景是,过去几年国家药监局批准上市的5.1类进口药的数量远远超过了1类创新药。另外,根据研发客统计,2021年国内发生的181笔授权许可交易中,从海外授权引进(License-in)新产品的交易首次破百,有105项,与2020年相比交易数量持续攀升。
观点
而从提高药品可及性、满足国内未满足临床需求的角度出发,将首先在国外上市而后再申请在中国境内上市的5.1类进口药,以及国内企业Licence-in模式下研发的药品排除在补偿制度之外,亦有可能对某些企业的积极性造成打击,无助于增加国内可用药品的多样性。
另外,参考国外已施行该制度的国家和地区的实践经验,也几乎没有在相关规定中去明确区分新药的“全球新”或者“地区新”,适用的对象都是“地区新”。
是“中国新”还是“全球新“?
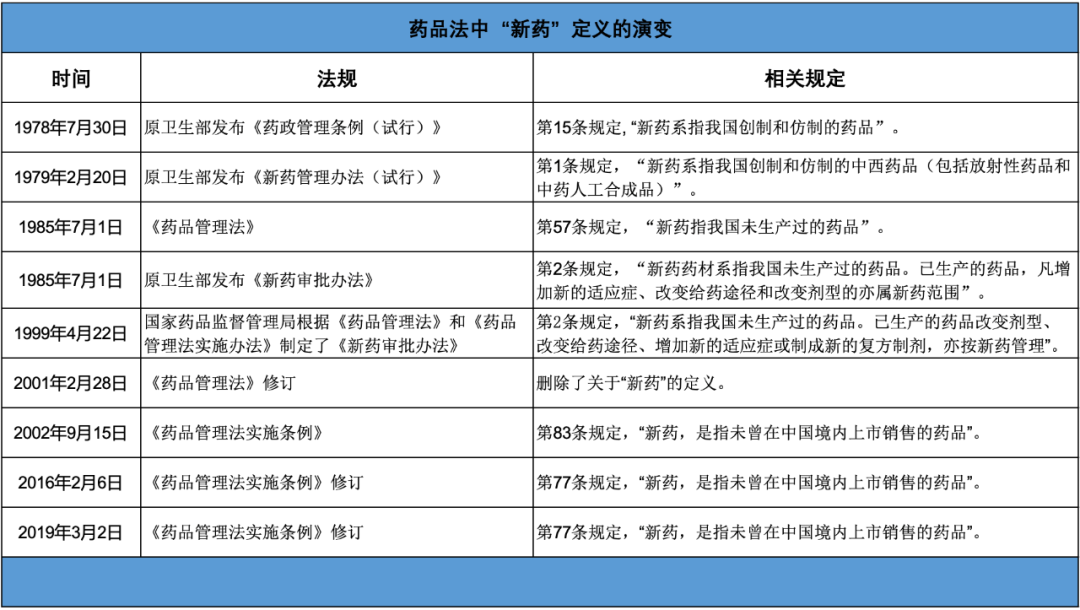
在《专利法实施细则》及《专利审查指南》的征求意见稿中,立法部门沿用了《药品注册管理办法》中曾出现的“新药申请”里的“新药”定义。《药品注册管理办法》在药品法的体系中属于部门行政规章,其上位法律和行政法规还有《药品管理法》等根本大法,以及《药品管理法实施条例》等行政法规。
观点
其实,纵观几十年来《药品管理法》和《药品管理法实施条例》的变化,并没有出现关于“中国新”还是“全球新”的争议。
如今再去看,如果需要定义的话,从1985年《药品管理法》中出现的“新药指我国未生产过的药品”到2019年的《药品管理法实施条例》中出现的“未曾在中国境内上市销售的药品”,“新”所包含的地域范围一直应该被理解为“中国新”。因而,即便是要对标药品法,“新药”的定义亦应该是“中国新”。
“新药申请”不应影响新药的定义
观点
值得注意的是,在2019年正式施行的《药品管理法实施条例》中,“新药”的定义依旧是“中国新”,立法者可能考虑到如果《药品注册管理办法》中“新药申请”使用“全球新”的概念,与其上位法《药品管理法实施条例》中“新药”的定义不符,因此,在2020年最终出台的《药品注册管理办法》中删除了关于“新药申请”的定义。
透过药品法中“新药申请”定义的变化,可以看出药品法立法者在有意区分“新药“和“新药申请”的定义,这种区分是合理的。“新药”更偏向实体法的概念,即什么样的药品、符合什么条件即可以被认定为“新药”。依照现行法律中依旧保留的定义,“中国新”应都属于“新药”。而“新药申请”更偏向于程序法,和药品注册审评审批流程紧密相关,在此过程中药监部门更关注的是药品安全性、有效性和质量可控性的审查。
对于未在国内上市的新药可能包括创新药、改良型新药、已在境外上市后寻求在中国境内上市的进口药,药监部门对这三类 “新药”采取不同的审评审批要求,适用不同的注册申请流程,是科学合理、符合实际的。但这种注册审评审批流程的区分从来没有,以后也不应该影响“新药”的定义,也不宜成为专利期补偿制度中“新药”定义对标的对象。







