

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

GPRC5D/CD3双抗:强生 vs 罗氏

GPRC5D是一种G蛋白偶联孤儿受体,在恶性浆细胞上过表达,正常组织的表达仅限于皮肤(毛囊和小汗腺)和睾丸(输精小管),且其表达水平与靶点BCMA相对独立。因此,GPRC5D作为下一个潜在治疗r/r MM的靶点备受关注。
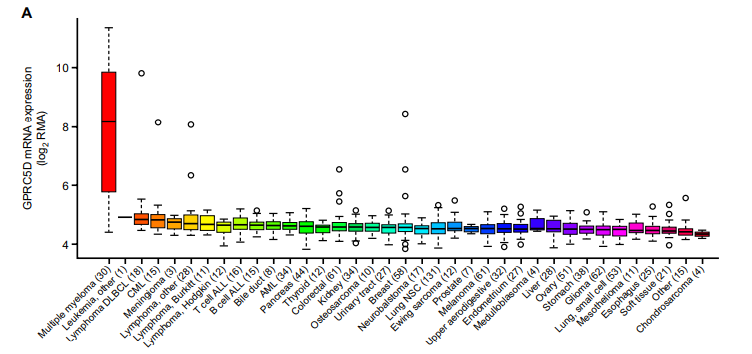
GPRC5D目前已有双抗、ADC、CAR-T细胞、CAR-NK细胞等药物形式出现。较为领先的是由强生开发的GPRC5D/CD3双抗Talquetamab(JNJ-64407564)和罗氏的RG6234,两款产品作用机制均是通过激活CD3阳性T细胞,诱导T细胞对GPRC5D阳性MM细胞进行杀伤。
本次ASH大会上,强生将公布I/II期MonumenTAL-1研究更新数据。截至2022年5月16日,288名之前未接受T细胞重定向疗法的患者在I/II期研究中接受了II期推荐剂量Talquetama治疗。143名接受0.4 mg/kg QW(每周一次)治疗患者的ORR为73%(≥VGPR: 58%;≥CR: 29%)。缓解程度随着时间的推移而加深。
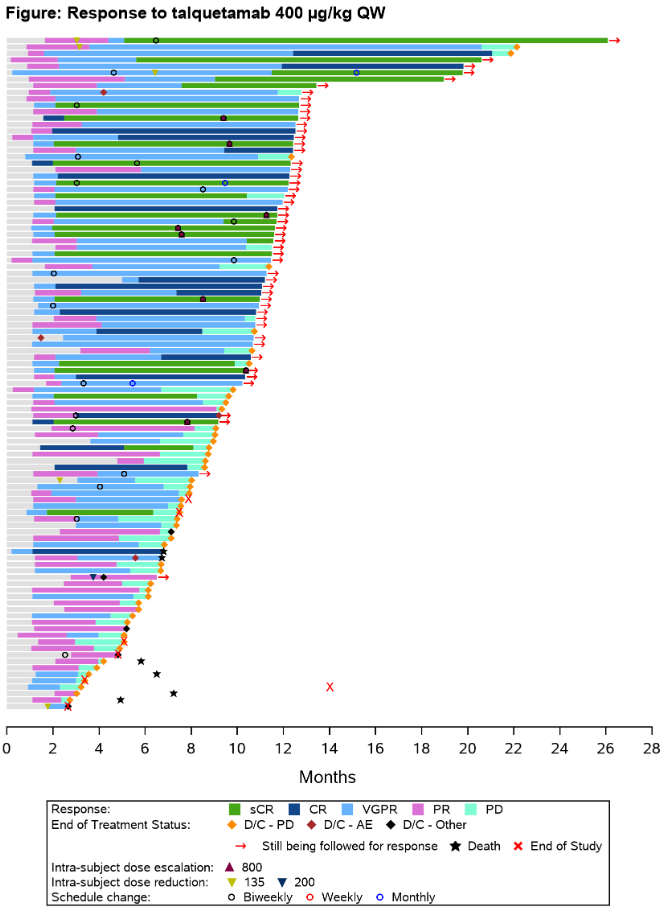
中位缓解持续时间(mDoR)为9.3个月(95% CI, 6.6-20.2)。中位无进展生存期(mPFS)为7.5个月(95% CI 5.7-9.2)。三重难治(72%[76/106])和五重难治性(71%[30/42])患者的ORR与总体人群相当。ASH会议上强生还将介绍0.8 mg/kg Q2W剂量组的疗效。
安全性方面,0.4mg/kg QW/0.8mg/kg Q2W最常见的AE为CRS (79%/72%;3级:2%/1%;4级:0%/0%),味觉障碍(48%/46%;3/4级:NA)和贫血(45%/39%;3级:31%/25%;4级:0%/0%]),皮肤相关的不良事件56%/68%(3级:0%/1%;4级:NA),指甲相关疾病52%/43%(3级:0%/0%;4级:NA),中性粒细胞减少34%/28%(3级:20%/17%;4级:10%/6%),血小板减少27%/27%(3级:10%/8%;4级:10%/8%),一般仅限于前几个周期。
罗氏公布了GPRC5D/CD3双抗RG6234的I期剂量递增研究静脉(IV)注射更新数据和首个皮下(SC)注射临床结果。
截至数据截止(2022年6月8日),已有51名患者接受IV治疗,54例接受SC治疗,IV组疗效评价患者中位随访时间为7.1个月(n=49例),SC组为3.9个月(n=48例)。ORR分别为71.4%和60.4%。
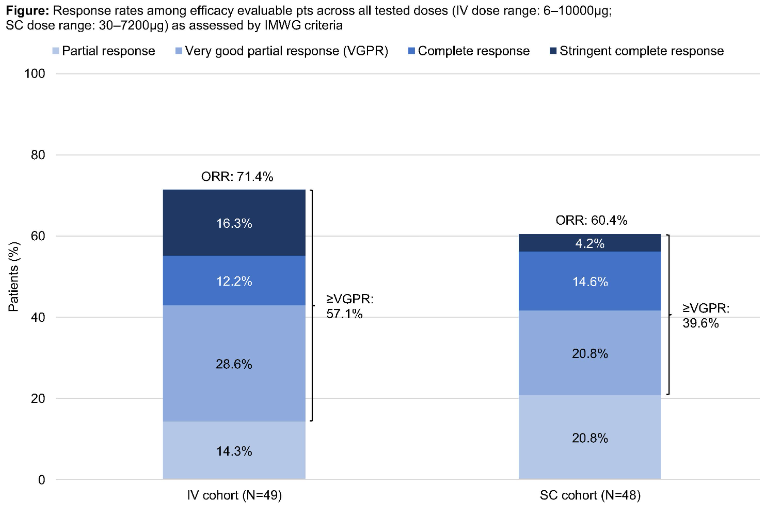
在接受过抗BCMA治疗的10/18名患者(55.6%)和有高危细胞遗传学的18/28名患者(64.2%)中观察到缓解。DoR数据在数据截止时不成熟。IV组24/35名患者持续缓解(68.6%),SC组26/29名患者持续缓解(89.7%),最大DoR分别为12.9个月和8.8个月。
CRS是最常见的不良事件(AE; IV: 82.4% ; SC: 77.8%),(Gr)≥3CRS(IV:2.0%;SC:1.9%)。与脱靶效应相关的AE包括对皮肤影响(IV: 72.5%;3级:11.8%;SC: 81.5%;3级:14.8%)、对头发和指甲的影响(IV: 17.6%;SC: 22.2%), 对胃肠道粘膜上皮或舌头影响(IV: 70.6%;SC: 74.1%;3级:5.6%)。Gr≥3的血液学不良事件包括,贫血(IV:13.7%;SC:5.2%),血小板减少(IV:13.8%;SC:18.5%);中性粒细胞减少(IV:11.8%;SC:16.7%),感染(IV: 56.9%;Gr≥3:19.6%;SC:37.0%;Gr≥3:24.1%)。IV组2名患者(3.9%)和SC组2名患者(3.7%)发生了RG6234相关不良事件,导致治疗终止。SC组报告1例(1.9%)RG6234相关Gr 5(致死性)急性呼吸衰竭AE。
CD38/IFNα抗体融合蛋白
在数据截止(2022年4月)时,100例患者接受了Modakafusp alfa治疗,56名在剂量递增组,44名在剂量扩展组。30名1.5mg/kg Q4W(每4周1剂)患者ORR为43%,mDoR未达到。mPFS为5.7个月。
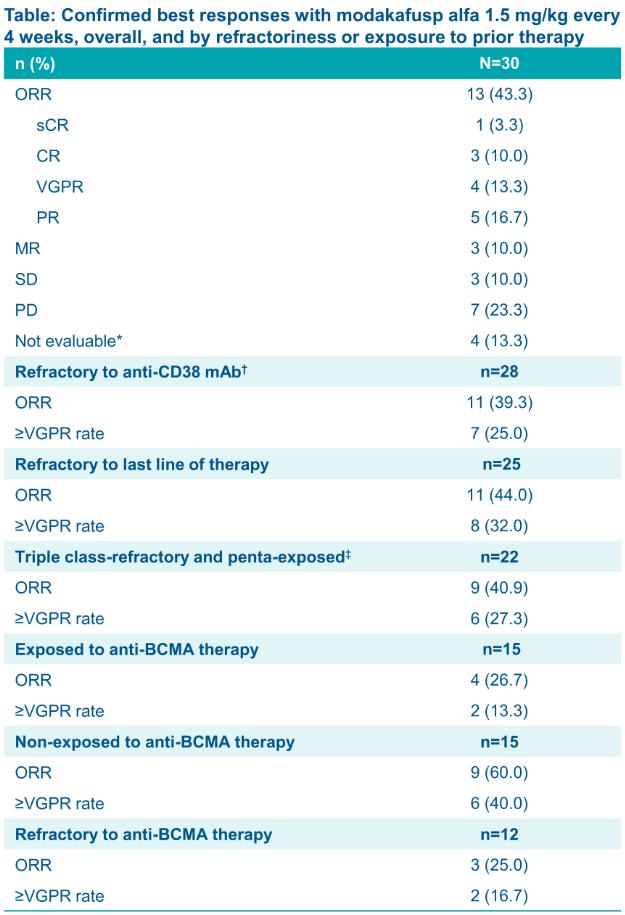
在1.5mg/kg Q4W组,26例(87%)患者发生了≥3级TEAEs,包括中性粒细胞减少症(n=19,63%[G4 :30%])、血小板减少(n=14, 47% [G4 :20%])和淋巴细胞减少(n=11,37%[G4:23%]);3例(10%)发生G3感染,1例(3%)发生G3出血事件。除1例报告G4高尿酸血症外,无G4非血液学TEAEs;11例(37% [G3=1,3%])发生了IRRs。
除了开发用于治疗MM,TAK-573还被开发用于治疗治疗转移性实体瘤。

CD3/FCRL5双特异性抗体
本届ASH年会上,罗氏将报告I期GO39775研究中,MM患者完成17个周期(C17)的cevstamab治疗后,停止治疗的早期缓解持续时间数据。
截至数据截止(2022年3月8日),共有16例患者完成cevostamab的C17处理,并符合分析条件。此前,这些患者接受了40-160mg的cevstamab,中位治疗周期为17(范围:16-17)。在16例患者中,最佳的总体缓解(BOR)是:7例严格完全缓解(sCR),3例CR,5例VGPR, 1例PR。
数据截止时,16例患者中的13例仍处于缓解状态,8例患者(BOR: 5 sCR, 1 CR, 2 VGPR)在治疗结束后维持6个月及以上的缓解,3例患者(BOR: 2 sCR, 1 CR)在治疗结束后维持12个月及以上的缓解。8例患者随访时间小于6个月。完成C17并获得sCR或CR的患者均未复发(0/10)。16例患者中仅有3例(BOR: 2例VGPR, 1例PR)在cevstamab C17疗程结束后发生PD。2例VGPR患者疾病进展时间分别为7.8个月和12.9个月。1例PR患者疾病进展时间为1.3个月。
口服分子胶降解剂
Mezigdomide是一种cereblon E3连接酶调节剂(CELMoD),与免疫调节剂(IMiD)相比,具有更强的杀灭肿瘤和免疫刺激作用。可诱导Ikaros和Aiolos的最大限度降解,使骨髓瘤细胞凋亡增加。临床前研究表明,MEZI与地塞米松(DEX)、蛋白酶体抑制剂(PI)和抗CD38单抗有协同抗癌作用。
C-92480-MM-001是一项正在进行的I/II期试验,旨在评估MEZI单药或联合DEX治疗RRMM患者疗效;通过I期剂量递增研究,将MEZI联合DEX的II期推荐剂量(RP2D)定为1mg,每日1次,持续21/28天。本届ASH年会上,BMS报告了MEZI+DEX在大量预处理r/r MM患者中的剂量扩展队列结果。
截至2022年5月24日,有101名患者在RP2D下接受了MEZI+DEX治疗,中位随访时间为5.8个月(范围0.5-19.0),23例(22.8%)患者继续接受治疗。停药的主要原因是疾病进展(50.5%)。
ORR为39.6%,其中2例(2.0%)达sCR,3例(3.0%)CR,18例(17.8%)达VGPR,17例(16.8%)达PR。虽然数据尚未成熟,但初步的mDoR为8.3个月(95% CI:5.4 -未达到),mPFS为4.6个月(95% CI 3.2-6.3)。浆细胞瘤患者ORR为30.8% (N=39),既往接受抗BCMA治疗患者ORR为50.0% (N=30)。
90例(89.1%)患者报告了Gr 3/4 TEAE。最常见(≥20%)的血液学Gr 3/4 TEAE为中性粒细胞减少症(74.3%,其中发热性中性粒细胞减少症14.9%)、贫血(32.7%)和血小板减少症(25.7%)。32.7%的患者发生3/4级感染;3/4期肺炎和COVID-19患者分别占9.9%和5.0%。72例(71.3%)和29例(28.7%)分别因TEAE而出现MEZI剂量中断和减少;6例(5.9%)患者因TEAT终止治疗,1例患者因血液学TEAE终止治疗。
癌症疫苗
SVN53-67是一种首创的新型肿瘤免疫治疗产品,其通过识别表达survivin蛋白的肿瘤细胞,并通过刺激患者自身的免疫反应控制肿瘤的生长和复发。复星医药拥有SVN53-67在中国大陆、香港及澳门的独家权益。
本次大会将公布一项I期试验的中期结果,评估由survivin肽偶联物SVN53-67/M57-钥孔傶血兰蛋白 (KLH) 组成的癌症疫苗方案在来那度胺(Len)维持治疗前后对MM患者的安全性和耐受性。
该研究纳入了年龄≥18岁的新诊断不符合或计划接受大剂量化疗和自体干细胞移植(HDC-ASCT)MM患者,并且他们在诱导治疗后至少达到部分缓解。
在分析时,18例患者接受了治疗,其中A组9例,B组9例。A组在第一剂疫苗接种4周后开始每日口服Len 10mg。B组在Len开始4周后接种第一剂疫苗。中位随访时间为41.5个月。共有18名患者接种了全部4剂疫苗。
在16/17(94%)患者中检测到对survivin肽内表位和相应野生型survivin肽的抗IgG抗体,A组和B组患者的效价无差异。其中10/17(59%)患者对survivin疫苗肽有高滴度(定义为>1:30 000)。在数据截止时,39%(7/18例)患者有疾病进展的证据,1例死亡。所有患者(A&B)的mPFS为24.8个月,mOS尚未达到。36个月的总生存率(OS36)保持在90.1% (95% CI, 50.8-98.7%)。
三特异性抗体
正在进行的I期研究为剂量递增阶段,旨在评估HPN217在r/r MM患者中的RP2D,PD/PK值,安全性以及初步疗效。
截至2022年6月27日,49例患者接受了HPN217治疗。目前的最高剂量水平为12000 μg/周,还将继续增加。最常见(≥20%)的TEAE为贫血(49%)、疲劳(37%)、CRS(25%)、恶心(22%)、关节痛(20%)、腹泻(20%)和转氨酶升高(20%)。所有CRS事件均为G1-G2(未报告≥G3事件)。未见免疫效应细胞相关神经毒性综合征(ICANS)的报道。在2860 μg/wk的浓度下有两种剂量限制毒性(DLT)。在逐步给药方案中没有DLT的报道,也没有达到MTD。
HPN217的PK值在不同剂量水平上呈现线性和比例升高,中位半衰期为66h (26-197h)。初始给药的一过性细胞因子升高更明显。达到客观缓解的患者在治疗期间可溶性BCMA降低、细胞因子升高,CD8+T细胞上活化标志物CD69上调。
BCMA单抗
Part A评估了SEA-BCMA单药剂量递增(100-1600 mg固定剂量静脉输注)和最高耐受剂量下的剂量扩展的疗效和安全性;此前的数据显示,SEA-BCMA160mg每2周一次 (Q2W) 通常耐受良好,显示出初始抗肿瘤活性,总缓解率 (ORR) 为14%(95% CI:2.9-34.9;n=22)。
本次ASH大会上,将公布SEA-BCMA强化给药(Part B)和加用地塞米松(DEX;Part C)的安全性、耐受性和临床活性。
截至2022年5月16日,共有73例患者接受了SEA-BCMA治疗。在Part B(n=15)、Part C1(n=12)和Part C2(n=8)中最常见的非血血学TEAE是疲劳(33%、50%和25%)、背痛(20%、17%和25%)、肺炎(7%、25%和25%)和咳嗽(20%、8%和25%)。最常见的血液学TEAE为贫血(C1:42%)。最常见的≥3级TEAE是贫血(C1:33%)和肺炎(B、C1和C2分别为7%、25%和0%)。
与Q2W单药治疗相比,强化SEA-BCMA给药(B)和添加DEX (C1)均显示ORR增加。Part B(n=15)的ORR为27%,mDoR为6.5个月。Part C1(n=12)的ORR为17%,mDoR尚未达到。
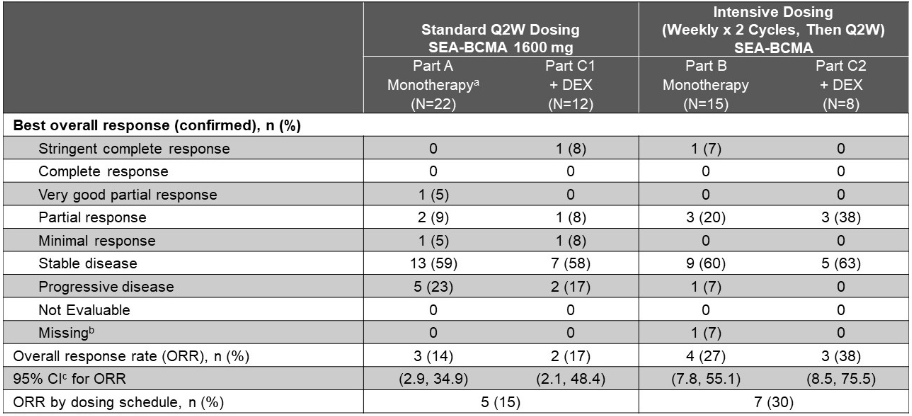
对于强化的SEA-BCMA给药方案和联合DEX(C2)组合,SEA-BCMA剂量水平(80mg和1600mg)的ORR为38%,mDOR尚未达到。两个强化给药组(B+C2)和两个标准给药组(A(1600mg)+C1)的综合ORR分别为30%和15%。
ASH2022相关阅读

医药魔方读者调研问卷
感谢陪伴 期待反馈
扫描二维码 提供宝贵建议







