病毒载体传播的基因转移方法是需要永久改变基因组的基因治疗方法的理想候选者。美国食品药品监督管理局(FDA)已批准基于LV的B细胞淋巴瘤疗法获得批准,而且基于LV的疗法被认为比γ逆转录病毒载体更安全。慢病毒载体还被证明可以转导最终分化的细胞,例如T细胞和神经元,从而扩大了它们的临床吸引力。
为了促进LV在临床上的广泛接受,慢病毒载体经历了重大的“一系列变化”,从而导致了致病基因元素的去除,并最终导致了第三代自我灭活(SIN)载体的发展[15]。第三代LV载体的独特设计特征是:(1)消除由长末端重复序列(LTRs)的U3区启动子序列引起的内在启动子活性,该序列位于野生型HIV的侧面,而仅通过内部启动子驱动转基因表达(2)删除负责自然HIV复制的tat基因(3)将病毒载体辅助元件分离为4个质粒[16]。慢病毒载体安全性的其他重大进步涉及生产系统的改进。
结果表明,只有三个必需基因(1)gag编码核心蛋白(2)pol编码病毒RNA结合蛋白,例如逆转录酶(RT),和(3)env编码病毒包膜蛋白,这些是产生功能性和高滴度病毒所必需的。
基于LV的递送用于治疗效果的临床应用的成功获得了针对包括癌症和单基因疾病在内的多种疾病的动力。这引起了诺华等大型医药公司的注意,该公司已获得了商业上使用慢病毒介导的递送系统来对抗细胞急性淋巴细胞性白血病(B-ALL)的批准。这突显了对基于基因治疗的临床应用的强烈需求,随之而来的是GMP级LV颗粒的制造需求的激增。
本文,作者描述了用于LV质粒制造的GMP方法,作者介绍已经纯化了约60 L的未浓缩上清液,最终体积约为200 mL。简而言之,他们的方案包括四质粒系统,使用扩增经主细胞库(MCB)认证的293T细胞瞬时转染。更换完全培养基,加入Benzonase处理以减少残留质粒的残留,过滤以去除细胞碎片并澄清病毒含有上清液的颗粒,通过Mustang Q色谱纯化。
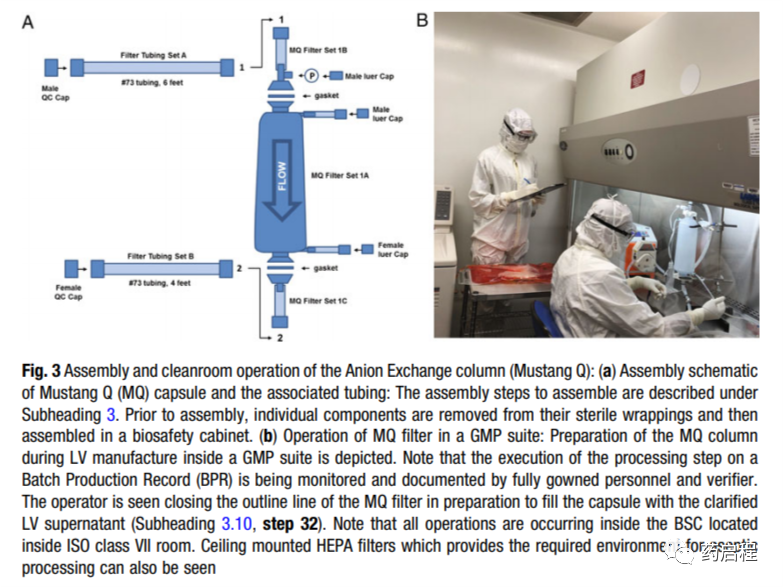
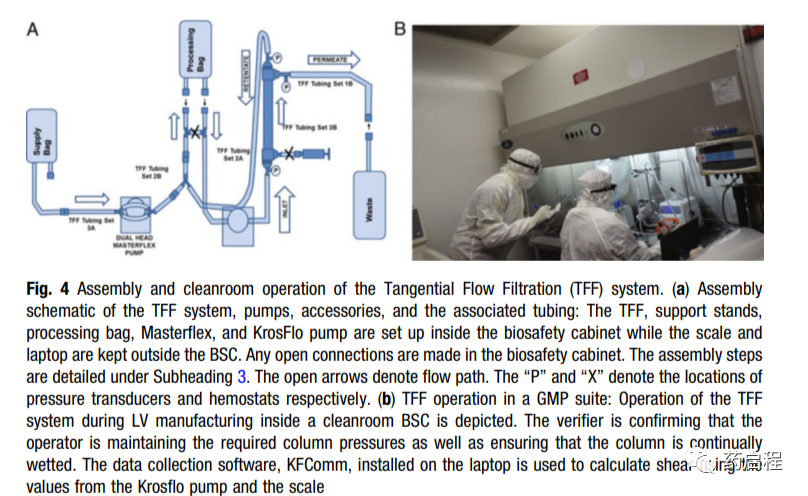
然后通过TFF过滤并渗滤到最终制剂缓冲液中。对于每种载体生产,应凭经验确定转染过程中所用质粒的量,收获时间以及色谱和过滤装置中所用缓冲液的离子强度和pH。
制造的质粒产品的放行标准中的测试项目,包括无菌性,内毒素水平,残留核酸/细胞和血清成分的测量以及任何复制型慢病毒(RCL)颗粒的检测。产品的滴度通常在0.5×108至5×108IU/mL之间。用该方案已经成功制备并认证了在美国,欧洲和亚洲用于临床的56种LV产品,其中大多数已经安全地纳入临床用途。
1、LV载体GMP生产中使用的FBS对采购和质量水平有不同要求,具体取决于将使用GMP LV的位置(美国,欧洲,亚洲)。建议在使用前与适当的监管机构核实这些要求。此外,由于不同批次的FBS可能会影响细胞生长,病毒滴度和产量,因此建议在购买并用于制造之前进行批次匹配并测试每批次的FBS。
2、建议将质粒商业化以确保高质量的制备。目前,不需要使用GMP级质粒来制备早期慢病毒载体。另外,用于LV载体的GMP生产的质粒的等级在监管机构之间可能不同,并且取决于产品开发的阶段。在使用前,质粒应通过测序确认其身份,无菌,内毒素水平低,DNA均一性大于95%,并且通常不含其制造过程中的杂质(细菌DNA)。
3、所有缓冲液均为商业购买的无菌产品,可立即使用,或者在经过认证的生物安全柜中制备,然后过滤0.22μm,所有使用的过滤器随后进行完整性测试。缓冲液应放入适当大小的实验室容器或生物处理袋中或转移至适当大小的实验室容器或生物处理袋中,以使液体能够被泵送。所有过滤器系统和管道组件也都组装在经过认证的生物安全柜中,然后包装并进行最终灭菌。
4、在放行LV之前,应先向监管机构和发起人核实放行产品所需的特定批次放行测试的标识。以下列表包含通常进行的测试,并应作为考虑的起点:无菌,内毒素,能复制的慢病毒(RCL)(细胞和上清液),支原体,体外分析外援病毒,载体滴度( 效价),p24的物理滴定度,残留的苯甲酸酶,载体插入序列,残留的细胞DNA,残留的质粒DNA,转染SV40 T抗原&腺病毒Type 5 E1a元件,残留的牛血清白蛋白(BSA),外观,pH,残留的宿主细胞蛋白和分子渗透压浓度。
识别微信二维码,添加生物制品圈小编,符合条件者即可加入生物制品微信群!


本公众号所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系(cbplib@163.com),我们将立即进行删除处理。所有文章仅代表作者观点,不代表本站立场。



 个人中心
个人中心
 我是园区
我是园区
 退出
退出







