从2015年9月高峰时期正在审评审批和等待审评审批的注册申请的近22000件降至2020年底的4882件。在极不平凡的2020年,CDE依旧很努力。
6
月
21
日,
国家药品监督管理局药品审评中心
正式发布《
2020
年度药品审评报告
》数据显示,
2020
年,
药审中心
全年审结注册申请任务整体按时限完成率为
94.48%
,其中临床急需境外已上市新药注册申请审结任务整体按时限完成率为
100%
,按默示许可受理注册申请的审结任务整体按时限完成率为
99.87%
,直接行政审批的注册申请
100%
在法定的
20
个工作日内完成,且审批平均用时
11.8
个工作日。
2020
年,药审中心完成了
13
个用于治疗罕见病的、临床急需的药品的技术审评,
且
均在规定时限内完成,罕见病药品在
3
个月之内完成审评,其他临床急需药品在
6
个月之内完成审评。
截至
2020
年
12
月
31
日,已发布的三批
81
个品种临床急需境外已上市新药中,已有
38
家企业的
48
个品种提出注册申请,其中
39
个品种已获批上市或完成审评,
100%
在时限内完成审评工作,
2020
年,
药审中心
完成
8606
件需技术审评的药品注册申请,
其中
化学药注册申请为
6778
件,较
2019
年增长
25.22%
;
中药注册申请
418
件,较
2019
年增长
39.33%
;
生物制品注册申请
1410
件,较
2019
年增长
27.72%
;
化学药注册申请约占全部技术审评完成量的
78.76%
。
在
8606
件需技术审评的药品注册申请中,完成新药临床试验(
IND
)申请审评
1561
件,较
2019
年增长
55.94%
;
其中
药审中心审评通过批准
IND
申请
1435
件,较
2019
年增长
54.97%
。
完成新药上市申请(
NDA
)审评
289
件,审评通过
NDA 208
件,较
2019
年增长
26.83%
,
药审中心审评通过创新药
NDA 20
个品种,审评通过境外生产原研药品
NDA 72
个品种(含新增适应症品种)
。
完成仿制药上市申请(
ANDA
)审评
1700
件
。
2020
年,药审中心加速推进仿制药一致性评价工作
,尤其是
在口服固体制剂一致性评价工作的基础上,积极推进注射剂一致性评价工作
,并在
2020
年
5
月份
正式启动注射剂一致性评价工作。
针对正式启动前已有
620
件待审评的注射剂一致性评价申请,药审中心成立专项审评工作组,采取细化分类处理措施,严格执行一次性发补,明确注射剂一致性评价注册检查的随机原则,加快审评速度,在不到
5
个月的时间内完成了
620
件品种的审评。
报告显示,
2020
年药审中心
完成仿制药质量和疗效一致性评价申请(以补充申请途径申报)
1136
件,较
2019
年增长
103.22%
,
审评通过批准一致性评价申请
577
件,较
2019
年增长
121.92%
。
其中,审评通过批准口服固体制剂一致性评价
456
件,审评通过批准注射剂一致性评价申请
121
件
。
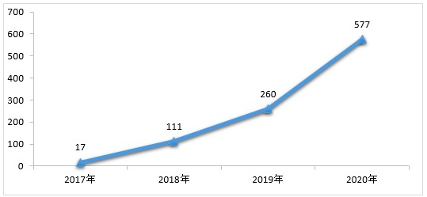
2018-2020年审评通过批准的一致性评价申请情况
2020
年,药审中心完成审评的化学药注册申请
6778
件。
其中,完成化学药临床申请(
IND
申请和验证性临床)共
1086
件,较
2019
年增长
45.58%
;
完成化学药
NDA 163
件;
完成化学药
ANDA 1697
件;
完成一致性评价申请
1136
件,较
2019
年增长
103.22%
;
完成化学药补充申请
2248
件,较
2019
年增长
23.72%
。
在
通过批准
IND
申请的
694
件
1
类创新化学药中,抗肿瘤药物、抗感染药物、循环系统疾病药物、内分泌系统药物、消化系统疾病药物和风湿性疾病及免疫药物较多,占全部创新药临床试验批准数量的
80.69%
。
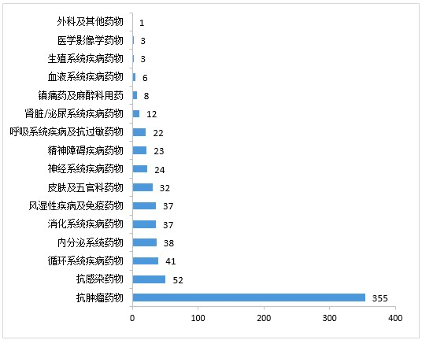
2020年审评通过批准的1类创新化学药IND申请适应症分布
数据显示,
2016-2020
年审评通过中药
NDA
的数量仍在个位数徘徊。
2020
年,药审中心完成审评的中药注册申请
418
件。
其中,完成
IND
申请
37
件,完成
NDA 8
件,完成
ANDA 3
件。
药审中心审评通过批准的中药
IND
申请
28
件,涉及
10
个适应症领域。
其中,呼吸
7
件、骨科
4
件、消化
4
件,共占
53.57%
。
而
2020
年
审评通过中药
NDA
也仅有
4
件
分别是,
连花清咳片、筋骨止痛凝胶、桑枝总生物碱片及桑枝总生物碱
。
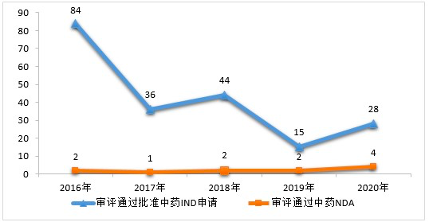
2016-2020年审评通过批准中药IND申请和审评通过中药NDA情况
值得关注的是,报告
2020
年因各类原因
导致审评结论为建议不批准的共
367
件
。
报告指出
药品注册申请在研发和申报过程中
存在的主要问题有:
在
新药申请
方面,
IND
申请和研发中存在的问题主要有:
正式申报前未开展沟通交流;
开发立题依据不足,成药性存在严重缺陷;
申报资料不足以支持开展药物临床试验或者不能保障临床受试者安全。
具体表现包括:
未沟通交流致使申报后发现研究信息严重缺项,无法在时限内完成补充研究;
已有的研究结果提示药效作用弱,毒性大,临床获益和风险比值不合理;
临床开发定位违背临床诊疗、用药的基本原则;
已有的药学、临床前研究不符合临床试验要求;
临床试验方案整体设计严重缺陷,风险控制措施不足;
联合用药的非临床研究数据不充分;
联合疫苗中单苗的数据不充分和
/
或免疫程序不一致。
NDA
研发和申报中存在的问题主要有:
研究质量控制和管理存在缺陷,导致已有的研究结果不能证明药品安全性、有效性和质量可控性;
违反合规性要求。
具体表现包括:
关键临床研究设计存在重大缺陷,无法得出客观、有力的有效性、安全性证据;
药学研究存在严重缺陷,无法证明产品的质量可控性;
各开发阶段的研究受试样品不一致;
注册核查中发现临床试验数据存在真实性问题。
在
仿制药一致性评价申请和上市申请中存在的问题主要有:
仿制药研发立题不合理;
申报资料无法证明仿制药与参比制剂(被仿制药品)的质量一致性。
具体表现包括:仿制药的参比制剂已撤市,且已有更新换代安全性更好的产品满足临床需求;样品复核检验不符合规定或分析方法存在严重缺陷;人体生物等效性试验结果表明不等效;样品稳定性研究结果、原料药起始物料选择等不符合仿制药上市技术要求;仿制药未按规定使用具有合法来源的原料药。
补充申请研究和申报中存在的问题主要有:
申请资料未能充分说明变更的科学性和合理性,不足以支持变更事项;
已有的研究结果不能保证变更后产品的安全性、有效性和质量可控性。
具体表现包括:
变更引起药用物质基础发生重大改变;
药品说明书修改申请不符合说明书撰写的技术要求;
用于支持变更的文献资料存在偏倚,或者临床安全性和有效性数据不充分。
生物类似药的开发方面,主要缺少相似性比较数据,药学比对研究中参照药选择存在缺陷;生物类似药临床前研究结果不足以支持其开展临床试验;天然药物的研究资料不符合国际多中心临床试验或我国天然药物评价基本技术要求。
文件显示,针对目前药品审评工作存在的问题,如
药审中心审评队伍规模结构与审评任务量配比失衡;
高层次及紧缺专业人才引进难、新进审评员急需长期专业培训等审评能力现代化短板问题;
以及
新旧注册相关规定过渡期间
的诸多问题,
2021
年药审中心
仍将
坚持鼓励药品研发创新
,
在创新药审评中探索实施
“
提前介入
”“
研审联动
”“
平行检验
”“
前置检验
”
等方式
深入推进监管科学研究,深化与高校、科研院所合作,加快首批重点项目研究成果转化
;
全面总结应急审评审批工作经验,完善审评审批制度体系,进一步激发药品创新发展活力。
值得关注的是,
加快建立符合中医药特点的中药审评机制体系
,也在重点工作之列,采取的措施有,将
组建古代经典名方中药复方制剂专家审评委员会,扎实推进中药审评审批改革;
参考
“
三方
”
审评审批经验,逐步探索适合古代经典名方的中药复方制剂的审评指导原则和标准,完善符合中医药特点的技术指导原则
等
。





 个人中心
个人中心
 我是园区
我是园区
 退出
退出








