

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

CDE 于 2021年9月16日发布《药物临床试验中心化监查统计指导原则(征求意见稿)》,泰格统计团队对此进行了学习和研读,结合当下国内热门讨论话题——远程智能临床试验和基于风险的监查等,进行知识点梳理,也结合泰格 RBQM 的研究成果提供一些建议。
为什么要中心化监查
近几年,临床试验的数据质量备受瞩目。数据质量的核心原则就是保护试验患者的权益以及保证最终分析数据的真实性和完整性,这也是 GCP 中的重点。
临床监查是提高试验数据的质量的重要措施,实现途径有多种,常见的有提高 SDV (Source Data Verification) 比例,在中国的临床试验,100%SDV 的比例+传统现场监查的方式一直是项目监查的执行标准。但是国外很多研究表明,SDV 比例高不一定就是数据质量好。Transcelerate 做过总结,对于整体数据质疑 (Query), SDV 的平均错误发现率只有平均 7.8%,关键数据的平均错误发现率为 2.4%。SDV 的比例解决不了全部的数据质量问题。
从 2016 年 ICH E6R2(章节5.0)发布了关于基于风险的监查(RBM Risk-Based Monitoring)的需求以来,FDA 在不断完善 RBM 策略。而 2020 年起,中国也开始全面实施 ICH 原则,并发布了新版 GCP(第31条),其中均明确提出采用系统的、基于风险的方式对临床试验进行监查。结合项目层面不同的情况采用不同的监查策略,中心化监查的方式既可以提高运行的效率也可以提高数据的质量。
中心化监查由申办方或者代表的各个职能部门协同从远程的方式对于项目数据( EDC 数据、运营数据等)及时对参与试验的中心进行评估。和传统的现场监查方式有所不同:
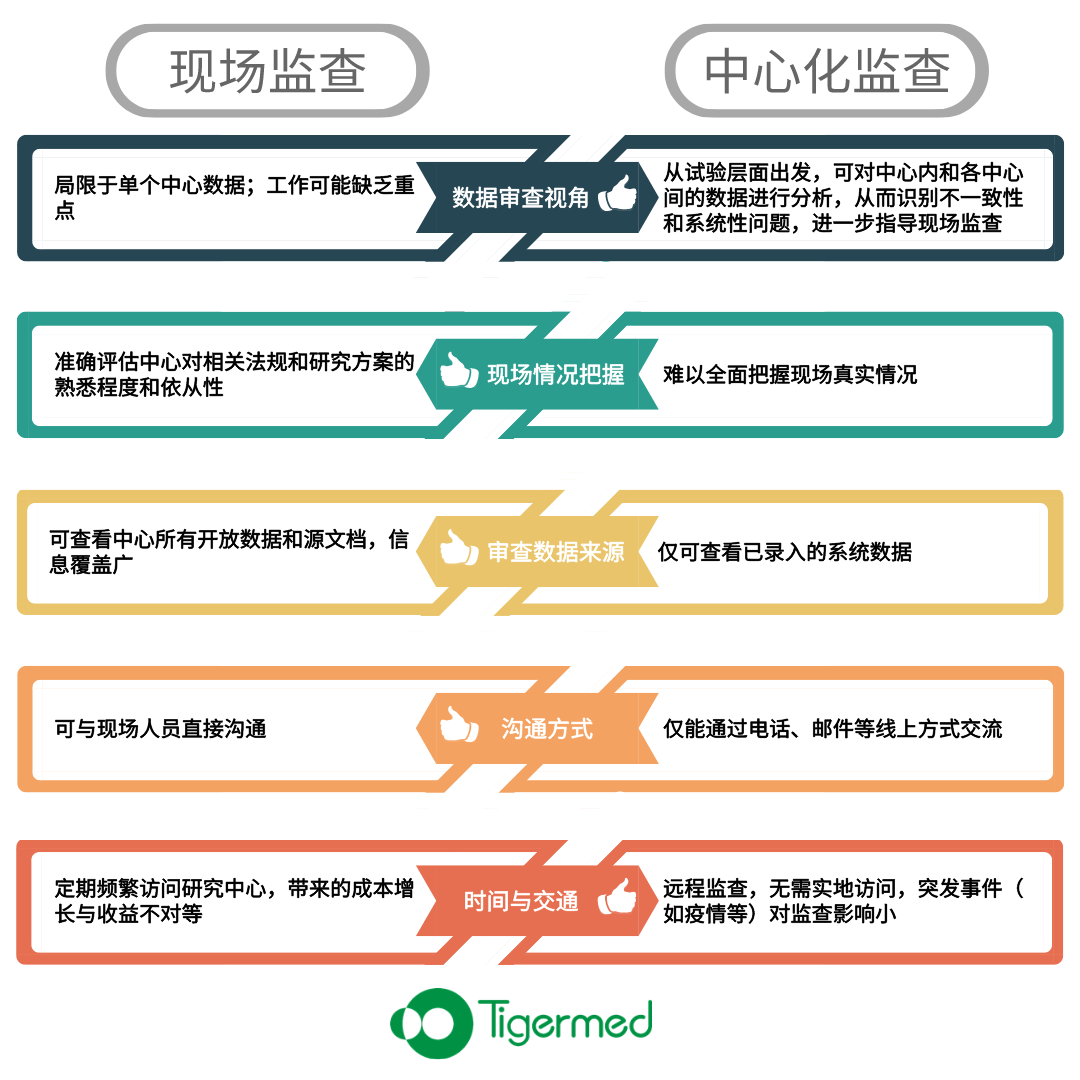
中心化监查不仅能作为现场监查的补充,还能帮助降低现场监查的频率和协助识别潜在问题数据,从而提示现场监查的重点。两者相互补充、相辅相成。建议采用两种监查方式相结合的模式,可提高临床试验的质量和效率,提高对患者权益的保护。
国内中心化监查的认知、技术研究和应用上还是早期阶段。作为中心化监查的重要内容:基于风险的质量管理 RBM/RBQM 的进展也备受瞩目。相对于国外的成熟度,DIA d-Trial 2021年1月在中国做的 RBM/RBQM 的调查问卷中,结果显示中国的 RBM/RBQM 大多在起步阶段。
CDE 发布的这个统计方法指南,利用统计学的科学性和方法论,对中心化监查中风险的识别和选择提供技术指导,表明我们国家在现代化临床试验中探索的坚定决心,也是未来临床试验的指南针和风向标,值得我们好好学习。
基于风险的质量管理体系的建立
我们常用“体检”来形容 RBM 的运行机制: RBM/RBQM 对进行中的临床试验周期性“体检”,可以帮助我们及时获知当前项目中关键数据和关键流程的整体情况。对于探测到的风险进行分析,有风险根据等级进行不同程度的降风险,根据趋势预测未来的风险,提高整体风险意识。
该部分内容较多,将在另外一篇中进一步解读。
中心化监查中的统计应用
统计方法和模型运用在分析临床试验的流程和临床数据,通过识别非典型的数据模式或者异常趋势实现。
3.1 常用统计指标
QTL: Quality Tolerance Limit 质量风险的容忍度。每个试验可以设定当前项目的 QTL , 也可以通用一些申办方或者试验药物层面的适用参数。用于检查受试者权益和试验数据完整性最相关的指标。根据项目情况和医学特征考虑影响受试者安全性和稳定性的指标,从整个试验的角度制定。比如:在高血压项目中,对于多次测量血压的结果计算数据的标准差,如果标准差 = 0 或 标准差 >= 4 就是“警告值”,在 >2 - <4 是“提醒值”,0< - <=2 是“正常值”。
(K)RI: (Key) Risk Indicator (关键)风险指标。KRI的制定更多是针对研究中心层面,通过统计量来判定一个中心的表现。Transcelerate 2019 年最新的风险指标列表推荐了 140 多种定性和定量的风险指标。项目层面挑选风险指标作为关键风险指标。风险指标很多,基于数据的可及性和可分析性,常见的 KRI 如下(不局限):
Monitoring report delays / 监查报告(MVR)延迟
Volume of SDV (Patient Visits)/ SDV 量(患者访视)
Volume of SDV (Safety Data Points) / SDV 量(安全性数据点)
Timeliness of data entry / 数据录入的及时性
Query response time (critical DP) / 质疑应答/答复时间(关键数据点)
Repeated values (Freq & Rate) / 重复值(频率&比率)
Monitoring Visit Report Approval Compliance / MVR 获批依从性
TMF completion / TMF 完成率
Below compliance criterion / 低于依从标准
Major protocol deviation / 主要方案违背
Number of informed consent issues / 签署知情同意书的问题数
AE rate / 不良事件发生率
Rate of discontinuation due to AE / 不良事件导致研究中止的比率
SAE timeliness / SAE上报时限
Screen failure rate / 筛选失败率
Subject discontinuation rate / 受试者终止率
质量容忍度(QTL)和关键风险指标(KRI)的有区别也有联系,常规的区别在于:
|
质量容忍度 (QTL) |
关键风险指标 (KRI) |
|
关注试验层面重要数据质量问题 |
关注多层面(尤其是中心层面)不良运营操作与数据趋势的风险 |
|
建议每个项目3-5 |
建议每个项目15-20 |
|
看试验整体数据,相对评估频率较少、时间较晚 |
累积一定数据量后开始定期监控 |
|
执行阶段不建议更改和调节阈值 |
执行阶段可根据试验需要调整KRI与阈值 |
|
需在CSR总结重要QTL偏离与补救措施,并向监管部门报告(ICH E3 章节9.6) |
无需向监管部门报告,内部管理使用 |
设定风险指标相应的风险阈值,在风险识别中,超出风险阈值触发相应的降风险措施。风险阈值的设定本身受试验目的、试验设计等因素影响,也和风险指标类型(定性、定量)、地区差异有关。风险指标的选择和阈值的确定需要统计团队和项目其他职能部门人员一起项目层面探讨确定。
3.2 统计方法
3.2.1
中心层面的 KRI 风险阈值用动态统计法和静态统计法
01
静态统计阈值法
静态阈值法是在 KRI 中各个中心都用统一的固定阈值进行风险判定。通常,业界公认的风险指标适用静态法。比如 SDV 比例,项目层面设定低于 80% 为高风险。
02
动态统计阈值法
动态阈值法,是按照项目整体数据的趋势来判定项目中各中心是否异常或者离群。比如,基于 3σ 原则,本文认为超过总体均值 2 倍和 3 倍标准误的值为异常值,相应的可以用 95% 和 99.7% 的置信区间定义控制限来判别异常值。下面列出几种动态统计阈值法:
1)离群值检测
如分位数法,在同一个项目层面,不同中心的方案违背率按照高低排序,高于 95% 分位点的设置为高风险。
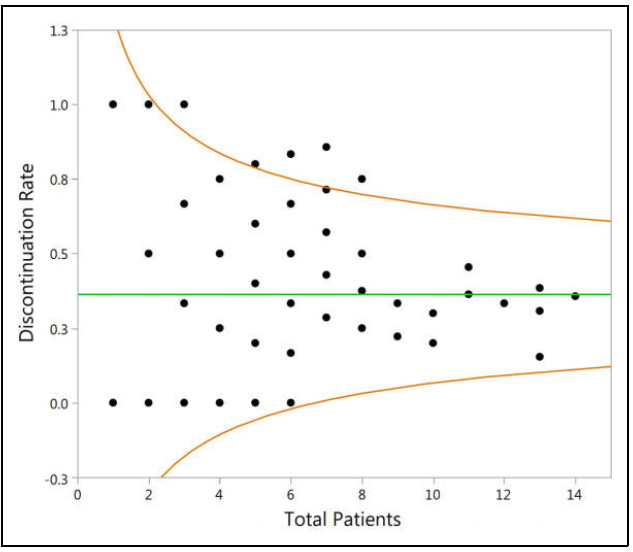
2)漏斗图(Funnel plots)
此方法适用于下列两种数据类型:二分类数据和计数型数据,分别基于二项分布和泊松分布进行统计计算:
二项分布:事件是否发生,二分类结果,比如提前退出率,源数据结果为是/否,漏斗图如下图:
泊松分布:适用于计数型数据,对于同一个受试者,事件可以多次发生,比如不良事件发生率,漏斗图如下图:
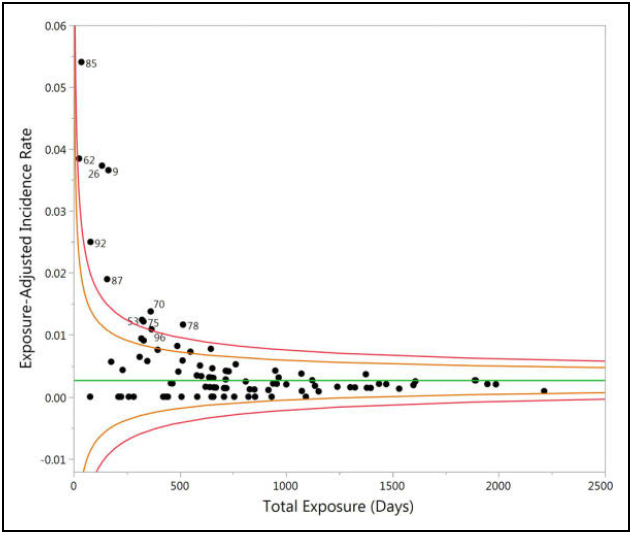
同时,试验中,中心的大小,或者中心启动后药物暴露的时间,对于不同的指标是有影响的。漏斗图法可以针对这些情况进行调整。
3)贝叶斯收缩法
对于计算次数的 KRI ,比如 AE 发生率,也可以用贝叶斯收缩法进行调整。
贝叶斯收缩估计量 = 整体估计值+ ( 1 - B ) * (本研究中心估计值- 整体估计值)其中B = 整体的估计精度/ (整体的估计精度+ 本研究中心的估计精度),其中估计精度的计算为1/估计方差。
即每家研究中心的估计量(后验)从实际观测值向整体均值(先验)进行不同幅度的“收缩”。方差大的中心,精度小,调整系数 B 大,更偏向总体估计值。
这里的精度需要知道中心层面的估计方差,如果 KRI 指标的数据没有波动,或者中心受试者人数很少,导致方差无法计算或者方差=0 的情况,起不到调整的作用。
4)基于模型的收缩法(探索中)
和贝叶斯收缩法相似,但是可以根据数据特征,估计解释风险因子的截距和斜率,用不同组合下的截距和斜率的置信区间的风险判定,多维度识别高危中心。
3.2.2
中心综合风险
结合多个指标,反映中心整体质量风险的单一评分:
权重法:根据经验预先给定每个 KRI 的权重,每个 KRI 的结果中高、中、低风险的等级对应不同的评分,用加权平均法得到总分。
主成分分析法(欧氏距离)
马氏距离等
评分衡量每个中心与研究整体数据分布的相似度。中心分数越高,距离越远,越离群,临床表现越异于其它中心。
3.2.3
试验数据的中心化监查
描述性统计方法或者图表支持数据趋势的探索或者异常值的发现。
Benford 定律检测数据的真实性等。
中心化监查的内容较多,本文罗列了统计指导原则相关的几点解读,后续会再发一篇关于基于风险的管理体系的建立。业界比较关注这个方向,方法的探讨还在持续。期待业界一起进行更多的探索和分享,推进中国化的中心监查道路。
1
END
1
作者:陈丽花







