

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

5月13日,EMA 发布了题为“关于质量受权人本人在场和个人住处征求意见”的文件,征求意见的时间为2022年5月13日-6月13日,该文件提供了关于质量受权人(Qualified Person,QP)远程认证或确认的问答,内容包括:
是否允许 QP 基于惯例进行远程批认证/批确认?
远程批认证/批确认应满足什么条件?
QP 必须是场地所在成员国的居民吗?
远程访问和批认证/确认使用签名的技术要求是什么?

图片来自网络
为防止药品短缺风险,同时确保药品质量、安全性和有效性,欧盟之前发布了“COVID-19 流行期间人用药监管期望问答”,对新冠疫情下的监管期望和灵活性给出了指导。这份文件对质量受权人可能允许的工作调整给出了回答,提到当现场检查不可行时,允许质量受权人进行远程认证,内容如下:
2.5 考虑到 COVID-19 大流行带来的旅行和其他限制,哪些质量受权人的工作可能可以调整?
如果已批准场地所在的成员国药监局接受,远程批认证/批确认是允许的。部分成员国药监局对 QP 的住址或实施远程批认证/批确认有特殊要求,生产商和 QP 应确认符合当地的要求。
应考虑以下要点:
完全符合欧盟立法和 GMP 要求;
批认证/批确认发生在 EU/EEA 范围内;
QP 在已批准场地花费的时间,应与该场地工艺相关的风险相称;
远程认证应在药品质量体系和相关的场地规程中进行描述和控制,对于允许合同 QP 的成员国,生产商和 QP 的技术协议中,应提及远程认证/确认,并明确 QP 必须在场的情况;
QP 做出批认证/确认决定时,应已电子获取相关必要信息,有未澄清的,或电子方式不能解决的问题或情况时,QP 必须亲自到场;
生产和进口许可(MIA)持有人应提供所要求的工具,使 QP 能够远程发挥功能,使用的 IT 系统应符合欧盟 GMP 附录11计算机化系统的要求;
对于 QP 远程实施的行动,批放行场地所在的监管当局应能同时获得,MIA 持有人有职责确保:a) 仅 QP 有批认证/确认和批次登记功能的入口;b) 传输的数据是完整且未经修改的;c) 有适当的电子签名系统。MIA 持有人应能证明,检查期间 QP 多久在现场一次,以及 QP 积极参与质量体系的监控。QP 必须能证明其履行了欧盟 GMP 附录16中的各项职责;
对于是否符合上述要点,应作为自检的一部分。
有些成员国可能有明确的国家要求。
最低要求应遵守欧盟 GMP 附录11计算机化系统。下列要求应进行调整,以反映当前技术的进展:
在将任何硬件异地转移之前,应对其进行识别和清点。应确保硬件保持完整和最新。硬盘应该加密,任何不需要的端口都应禁用;
对于可能使用虚拟专用网络的 QP,应适当配置网络操作系统、数据库和应用程序级别上的安全参数,以避免未经授权的访问;
认证和授权应使用公认的行业标准(例如双因素或多因素身份验证)。不应使用共享的身份验证信息,应有身份验证信息的自动过期;
传输中的数据应通过强传输加密(例如TLS 1.2、https)进行保护;
MIA 持有人负责实施组织控制(例如个人权限的分配)和技术控制,以确保只有 QP 能够执行远程批认证/确认。
欧盟法令2001/83/EC 第51条规定了 QP 的职责(兽用药对应法令2001/82/EC 的第55条),欧盟 GMP 附录16 药品质量受权人签发证书和批放行中提出了更多详细要求:
1.6 QP 必须亲自确保在认证批次可放行至市场或出口之前履行以下操作责任:
1.7 此外,QP 应负责确保1.7.1至1.7.21条的内容。可将这些任务委托给经过适当培训的人员或第三方。公认的是,QP 需依靠药品质量体系,并且 QP 应持续保证依靠药品质量体系是有根据的。
1.7.1 与药品生产和检验相关的所有活动均按照 GMP 的原则和指导进行。
1.7.2 原料药和药品直至认证前的整个供应链均已记录在案 , 并可供 QP 查询。这应包括药品的起始物料和包装材料的生产场地 , 以及通过生产工艺风险评估被认为是关键的任何其他物料的生产场地。记录文件最好应采用综合图表的格式 , 其中应纳入所涉各方 , 包括无菌加工的成分和设备的灭菌等关键步骤的分包商。
1.7.3 已经对药品生产和检验所涉场地以及原料药生产所涉场所进行了全部审计,并且审计报告可供执行认证的 QP 使用。
1.7.4 所有生产、分析和认证场地均符合目标区域的 MA 条款。
1.7.5 所有生产活动和检验活动均与 MA 中描述的一致。
1.7.6 批次中使用的起始物料和包装材料的来源和质量标准均符合 MA 的要求。供应商具备质量管理体系,能确保仅供应所要求质量的物料。
1.7.7 对于属于经修订的第2001/83/EC 号指令或第2001/82/EC 号指令范围内的药品 , 其原料药是按照 GMP 生产的,并在需要时按照原料药经营质量管理规范(GDP)进行销售。
1.7.8 用于人用药生产的原料药的进口应符合经修订的第2001/83/EC 号指令第46(b)条的要求。
1.7.9 对于属于经修订的第2001/83/EC 号指令范围内的药品,其辅料是按照该指令第46(f)条中确定的 GMP 进行生产的。
1.7.10 在相关时,用于批生产的所有物料的 TSE(可传播性海绵状脑病)状态均符合 MA 的规定。
1.7.11 所有记录都是完整的,并由相关人员认可签字。已进行所有要求的中间过程控制和检查。
1.7.12 所有生产和检验程序均保持经验证状态。人员酌情接受培训和资质确认。
1.7.13 成品质量控制(QC)检验数据符合 MA 或经许可的实时放行检验计划中的成品质量标准。
1.7.14 已解决所有与药品生产和检验有关的注册上市后承诺。持续稳定性数据一直能支持认证。
1.7.15 已经评估了任何变更对药品生产和检验的影响 , 并且所有其他检查和检验均已完成。
1.7.16 与被认证批次有关的所有调查(包括超标和超趋势的调查)的完成程度足以支持认证。
1.7.17 正在进行的任何投诉、调查或召回均不会使该批次的认证条件无效。
1.7.18 所需的技术协议均已准备就绪。
1.7.19 自检计划处于有效状态且是现行的。
1.7.20 分销和发运的适当规定已准备就绪。
1.7.21 对于拟在欧盟市场上销售的人用药品,经修订的第2001/83/EC 号指令第54(o)条所指的安全特征已贴在包装上(如适用)。



大会议程
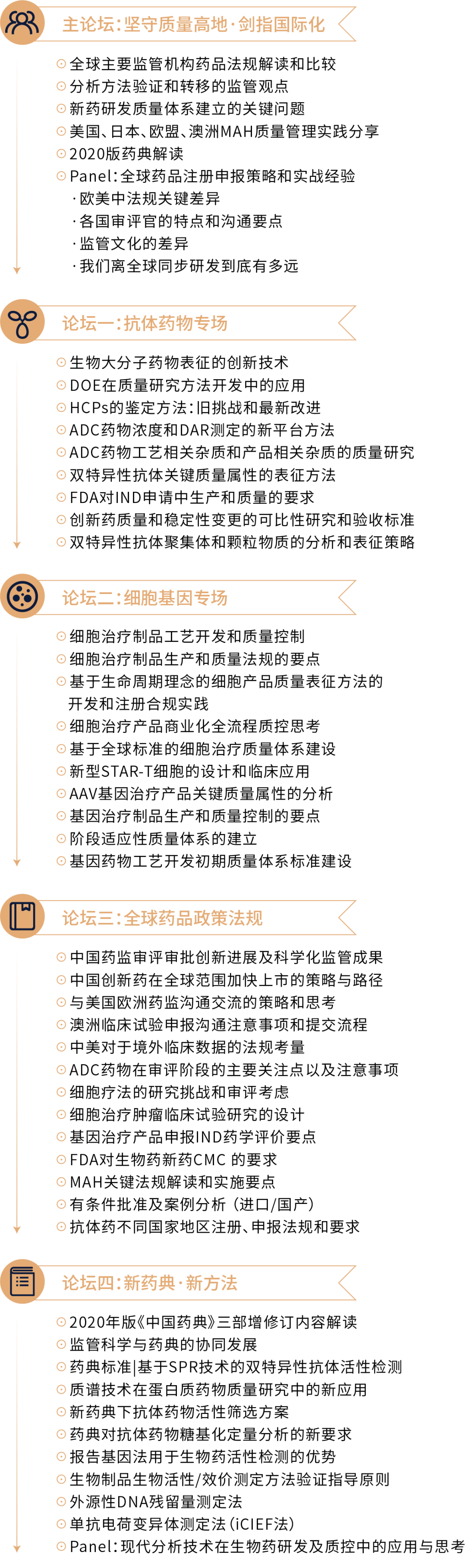
大会亮点
1.
国内首个:
聚焦生物药质量和政策法规领域的千人大会
2.
连续三年:
药品质量理念和专注态度的延续,兼具深度和广度,全新再出发
3.
剑指国际化:
解读创新药国际注册法规和质量要求,理解各国监管文化,制定全球战略
4.
云集一流:
顶尖制药巨头、创新型药企、监管部门等的重量级嘉宾
5.
理论+实战:
从分析方法研发到质量管理,理论结合实战,提供思路与方法,解决实际问题
参会报名
❖ 药企人员限时免费注册中

【扫码立即报名】
❖ 定制参展/大会报告/参会报名/媒体合作请联系:
联系人:Abby
手 机:18217659261(微信同号)
邮 箱:xiaolang.jiang@biovalleyclub.com

【备注:QbD,进入质量大会群聊,实时共享大会及行业前沿信息】






