

 个人中心
个人中心
 我是园区
我是园区
 退出
退出
您还不是认证园区!
赶快前去认证园区吧!

据 Insight 数据库统计,本周(1 月 1 日-1 月 7 日)全球共有 47 款创新药(含改良新)研发进度推进到了新阶段,其中 1 款获批上市,4 款申报上市,10 款获批临床,15 款申报临床。
下文中,Insight 将分别摘取国内外部分重点项目做介绍。

国内创新药进展
国内部分,本周共有 30 款创新药(含改良新)研发进度推进到了新阶段,其中 1 款获批上市,1 款申报上市,9 款首次启动临床,14 款申报临床。
本周国内首次启动临床的 9 款创新药(含改良新)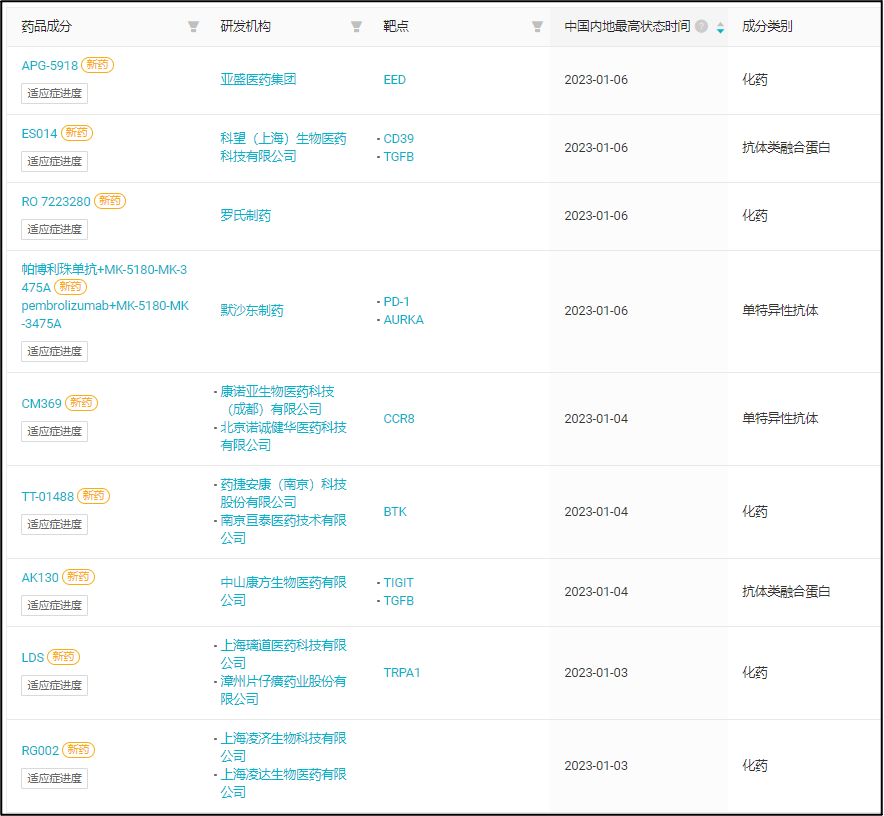 来自:Insight 数据库网页版(http://db.dxy.cn/v5/home/)
来自:Insight 数据库网页版(http://db.dxy.cn/v5/home/)
获批上市
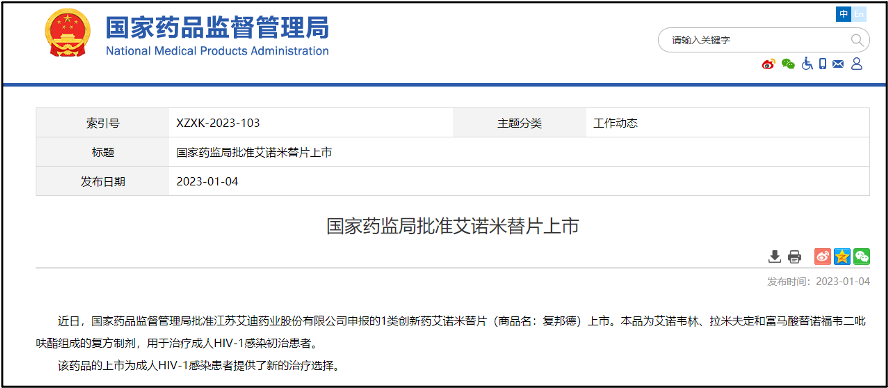
艾迪药业:开门红!「艾诺米替片」获批上市
1 月 4 日,NMPA 发布公告,批准江苏艾迪药业股份有限公司申报的 1 类创新药艾诺米替片(商品名:复邦德,研发代号:ACC008)上市。这是一款艾诺韦林、拉米夫定和富马酸替诺福韦二吡呋酯组成的复方制剂,用于治疗成人 HIV-1 感染初治患者。同时值得一提的是,这也是 2023 年度 NMPA 批准的首款 1 类新药。
此前在 2021 年 6 月,艾诺米替的复方成分之一、1 类新药艾诺韦林(研发代号:ACC007)也已经获 NMPA 批准上市。
艾诺韦林是全新结构的非核苷类逆转录酶抑制剂,通过非竞争性结合并抑制 HIV 逆转录酶活性,从而阻止病毒转录和复制。这是艾迪药业的首个抗艾滋病 1 类新药,被列入国家十三五「重大新药创制」科技重大专项。
2016 年 8 月,艾迪药业提交艾诺韦林的新药临床试验申请,2017 年 3 月取得了临床试验批件;I 期临床试验结束后经国家药品审评中心同意,于 2018 年 7 月被批准可直接进入 III 期临床试验阶段。
III 期临床试验主要包括一个多中心、随机、双盲双模拟、阳性平行对照、非劣效临床试验,主要目的为证明在未经抗逆转录病毒药物治疗的 HIV 感染者治疗 48 周时,艾诺韦林试验组有病毒学反应(病毒载量小于 50 拷贝/毫升)受试者的比例不劣于依非韦伦对照组。
结果显示,艾诺韦林达到了主要临床终点指标,和对照组相互非劣等效,在不良反应尤其是各类神经系统和精神类不良事件的发生率方面则优于对照组。
而抗艾滋三联单片复方制剂艾诺米替片,即本周获批新药,是艾迪药业在艾诺韦林的基础上,联合 2 个核苷类逆转录酶抑制剂(拉米夫定和替诺福韦)开发的国产首款三合一单片复方创新药制剂,其组合方案及药物选择符合国际趋势,于 2019 年 12 月被列入国家十三五「重大新药创制」科技重大专项。
对于艾诺米替,HIV 患者每天仅需服用 1 片,无需再服用其它抗艾滋病药物,有助于减轻患者服药负担,增加依从性,减少耐药发生,可为国内患者提供一个国际同步的新选择。
2021 年 3 月,艾诺米替片的生物等效性试验顺利完成,5 月份,艾迪药业以与 ACC007 相同的适应症申请上市,本周终于获批上市。此外,艾诺米替片针对 HIV-1 经治患者的 III 期临床研究方案也已通过临床研究中心组长单位北京地坛医院的伦理审核,相关临床研究工作正在有序推进中。
申报上市
亿腾景昂:
针对乳腺癌,1 类新药「恩替诺特」申报上市
1 月 4 日,亿腾景昂药业 1 类新药恩替诺特首次在国内申报上市。这是一款选择性组蛋白去乙酰化酶(HDAC)抑制剂,用于治疗激素受体(HR)阳性、人类表皮生长因子受体 2(HER-2)阴性晚期乳腺癌患者。
恩替诺特最早由拜耳先灵制药开发,原代号为 MS-275,2007 年 4 月转让给 Syndax Pharmaceuticals 公司。后续在 2013 年和 2015 年,Syndax 公司又将大中华区及泰国、马来西亚和新加坡的独家权益转让给亿腾景昂,将日本、韩国权益转让给协和麒麟。
此前这款新药的全球最高状态处于临床 3 期,在 2021 SABCS 上,亿腾景昂发布了针对中国乳腺癌患者的 III 期临床结果(ClinicalTrials.gov 登记号:NCT03538171)。亿腾景昂是各权益方中首个将其推入上市申报流程的企业。恩替诺特全球首项临床登记发生于 2004 年,迄今已经历时 18 年研发。
据 Insight 数据库显示,亿腾景昂在引进这款新药之后,2015 年 3 月递交了临床申请,2016 年 7 月启动首项临床试验。在国内目前仅开发了乳腺癌适应症,不过全球范围来看,恩替诺特布局的领域还包括血液系统肿瘤(霍奇金淋巴瘤、急性淋巴细胞白血病、慢性髓系白血病等)、结直肠癌、胆管癌、输卵管癌、腹膜癌等,曾探索的联用组合包括 K 药、T 药、恩扎卢胺等。
不过,此前该药也曾在多项 I/II 期和 III 期临床的探索中遇到挫折,如联合 T 药阿替利珠单抗治疗乳腺癌和结直肠癌的早期临床、联合 K 药帕博利珠单抗治疗非小细胞肺癌的早期临床、以及联合依西美坦治疗乳腺癌的 III 期临床。
恩替诺特是一款 HDAC 抑制剂。在国内,已上市的同靶点药物为西达本胺,其余还有 4 款 III 期临床,不过 III 期临床产品中每款适应症布局都有所差异。而几款中国处于 III 期临床的 HDAC 抑制剂,适应症布局对比如下图:
4 款国内 III 期临床 HDAC 抑制剂适应症对比
 来自:Insight 数据库网页版
来自:Insight 数据库网页版
信达生物:
PI3Kδ 抑制剂上市申请获受理,已纳入优先审评
1 月 6 日,信达生物/Incyte 公司合作开发的 PI3Kδ 抑制剂帕萨利司片(Parsaclisib)已递交上市申请并获得 CDE 受理。此前在 2022 年 10 月 25 日,Parsaclisib 已经在 CDE 拟优先审评名单中公示,现已正式纳入优先审评通道,用于既往接受过至少两种系统性治疗的复发或难治性滤泡性淋巴瘤成人患者。
Parsaclisib 来源于信达 2018 年度与 Incyte 公司的合作。在该项合作中,信达生物获得了 3 款抗肿瘤药物在中国内地及港澳台地区的临床开发和商业化权益,而 Incyte 公司则获得信达生物的 4000 万美元首付款、首次递交 IND 申请后的第二笔 2000 万美元付款、潜在开发及监管里程碑款最高 1.29 亿美元和潜在商业里程碑 2.025 亿美元。
这 3 款药物分别为:itacitinib(JAK1 抑制剂)、pemigatinib(FGFR 抑制剂)和 parsaclisib(PI3Kδ 抑制剂)。在国内,佩米替尼(pemigatinib)已经于 2022 年 4 月获批上市,如今 parsaclisib 紧随其后也递交了上市申请,itacitinib 则处于 1/2 期临床中。
医药交易详情
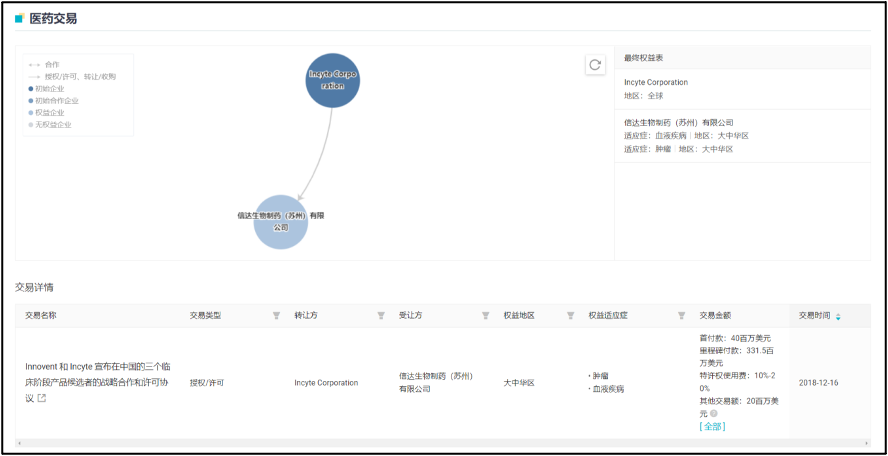 来自:Insight 数据库网页版
来自:Insight 数据库网页版
信达生物在 2022 ASCO 会议上发布了 Parsaclisib 在中国人群中的数据:在滤泡性淋巴瘤患者(N = 61)中 ORR 达到 86.9%(95%CI,75.8-94.2),其中 31.1%(95%CI,19.9-43.3)达到 CR:
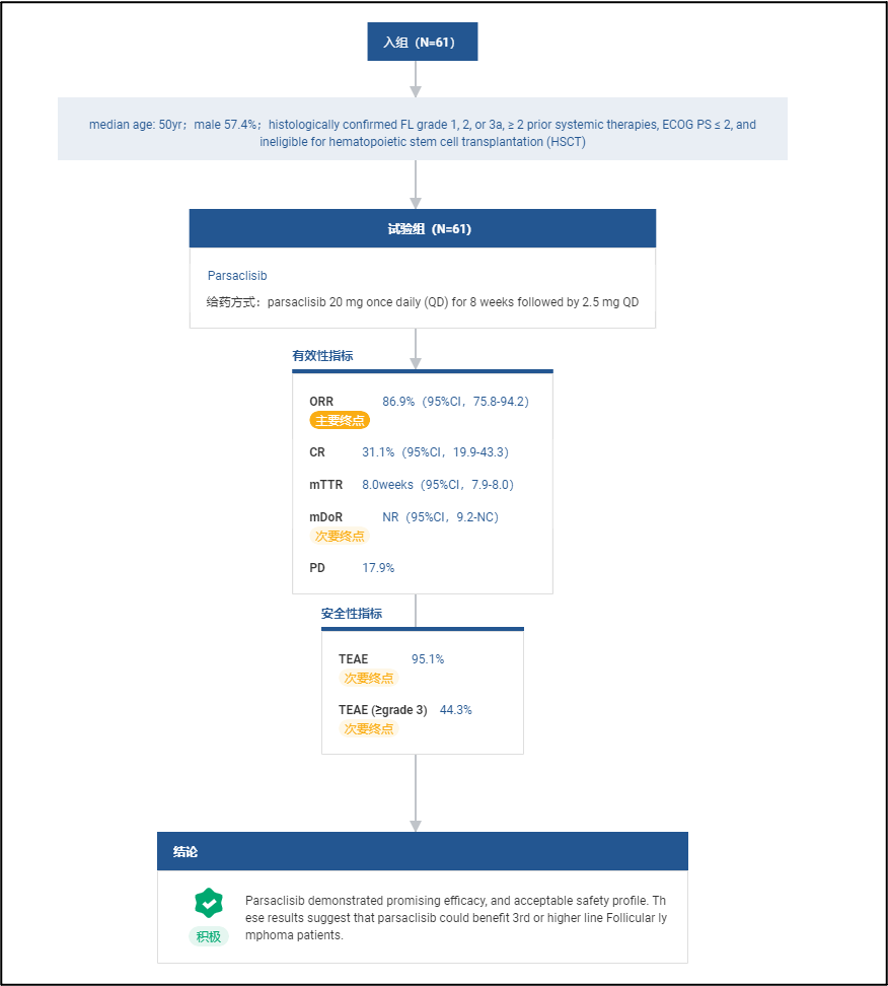 来自:Insight 数据库网页版
来自:Insight 数据库网页版
国内来看,Parsaclisib 是第 3 款申报上市的 PI3Kδ 抑制剂。2022 年 3 月,石药引进的 PI3Kγ/δ 抑制剂度恩西布已经获批用于滤泡性淋巴瘤;恒瑞和璎黎合作的林普利塞自 2021 年 5 月申报上市,2022 年 11 月已经获得批准。
国内临床 II 期及以上 PI3Kδ 抑制剂
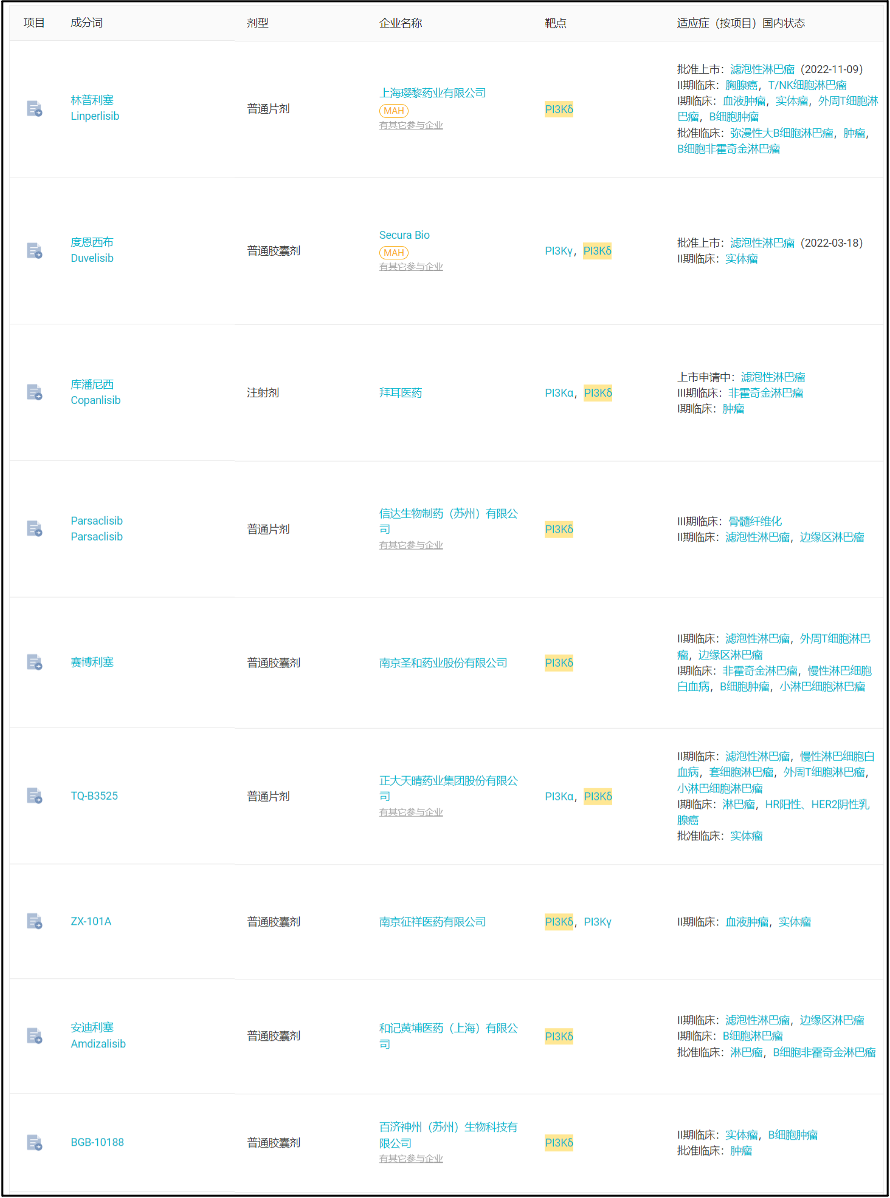 来自:Insight 数据库网页版
来自:Insight 数据库网页版
申报临床
海思科:
EGFR 靶点新变局!国内首款 PROTAC 新药报临床
1 月 4 日,CDE 官网显示,海思科 HSK40118 片首次在国内申报临床,拟用于治疗非小细胞肺癌。值得注意的是,这是国内企业首款申报临床的 EGFR PROTAC。
HSK40118 片是海思科自主研发的口服 EGFR-PROTAC 小分子抗肿瘤药物,是基于海思科领先的 Protac 研发平台筛选出的第 2 个小分子抗肿瘤药物,由靶向 EGFR 蛋白的小分子抑制剂、E3 泛素连接酶的招募配体和连接这两个部分的 linker 组成的三联体。临床拟用于治疗 EGFR 突变的晚期非小细胞肺癌。
肺癌是中国高发病率的癌种之一,其中非小细胞肺癌占比约为 85%;而在非小细胞肺癌患者中,EGFR 又是最为常见的基因突变类型,约占 50%。
EGFR-TKI 是治疗此类突变最有效的药物。2016 年-2020 年,中国 EGFR 小分子靶向药物市场从 22 亿元增长至 108 亿元,随着基因检测技术的不断完善和普及,预期到 2025 年,中国 EGFR 小分子靶向药物市场规模将达到 368 亿元,针对非小细胞肺癌患者更有效的靶向治疗方案,仍然存在大量未被满足的临床需求。
目前 EGFR-TKI 已经从第一代发展至第四代,而第三代 EGFR-TKI 的竞争无疑最为激烈,除了首先获批的 3 款已进医保的新药奥希替尼(阿斯利康)、阿美替尼(翰森制药)和伏美替尼(艾力斯)之外,第二梯队的 4 款贝福替尼(贝达药业)、瑞泽替尼(石药集团/倍而达)、Limertinib(奥赛康)和奥瑞替尼(圣和药业)也都申报上市。
在第三代之后,针对 EGFR 罕见突变的 EGFR-TKI、EGFR/c-Met 双抗研发已在路上。海思科开发的 HSK40118 片虽暂未披露主要靶向的 EGFR 突变类型,但作为国内首个同靶点 PROTAC,后续临床开发值得关注。
海思科是国内 PROTAC 领域布局领先的企业之一。目前据 Insight 数据库显示,由中国企业研发的 PROTAC 药物中,进入临床阶段(IND 申请及以上)的仅有 6 款,其中 2 款来自于海思科。该公司的首款 PROTAC 靶向 BTK,研发代号为 HSK29116,2021 年 4 月已经进入 I 期临床。
由国内企业开发、已启动临床试验的 5 款 PROTAC
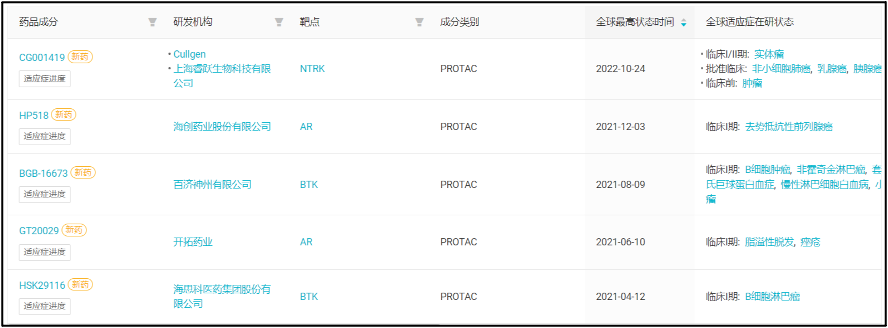 来自:Insight 数据库网页版
来自:Insight 数据库网页版
临床试验结果披露
康希诺生物:
新冠病毒 mRNA 疫苗 3+1 序贯数据公布,更安全效果好
1 月 5 日,康希诺生物发布公告,与下属公司康希诺(上海)生物科技有限公司共同开发的新型冠状病毒 mRNA 疫苗 CS-2034 在一项评估序贯加强安全性和免疫原性的临床研究中,取得了积极的阶段性数据。
康希诺生物新型冠状病毒 mRNA 疫苗 CS-2034(以下简称「CS-2034」)是针对现有新冠病毒变异株有更好保护效果的新一代疫苗,可以诱导出针对多种世界卫生组织认定的奥密克戎等重要变异株的高滴度中和抗体,具有更强的广谱性,可以更有效地保护机体免受现有变异株的感染。目前,该疫苗处于临床 IIb 期阶段。近日,该疫苗在安全性和免疫原性临床研究取得了积极的阶段性进展。
研究表明,已接种 3 剂灭活疫苗的 18 岁及以上人群,采用康希诺生物新型冠状病毒 mRNA 疫苗进行序贯加强具有良好的耐受性,针对奥密克戎 BA.5 变异株的中和抗体水平是灭活疫苗同源加强的 29 倍。60 岁及以上的老年人群接种安全性更佳,可获得较强的免疫保护。
该临床研究分 A、B 两组,A 组受试者分为 18-59 岁和 60 岁及以上 2 个年龄层,其中 60 岁及以上的老年人占比高达 50%。受试者按照 3:1 的比例随机接种 1 剂 CS-2034(以下简称「mRNA 组」)或新冠灭活疫苗(以下简称「灭活组」),其中 mRNA 组接种疫苗为 0.3 ml,灭活组接种疫苗为 0.5 ml。B 组均为 60 岁及以上的老年人,观察该组受试者在接种 1 剂 CS-2034 后的安全性。
安全性结果显示,在既往接种过 3 剂灭活疫苗的人群中序贯加强接种 1 剂 CS-2034,安全性良好,不良反应发生率及严重程度显著低于文献报道的已上市 mRNA 疫苗。值得注意的是,60 岁及以上老年人组的安全性优于 18-59 岁成年人组,为老年人群加强新型冠状病毒 mRNA 疫苗提供了强有力的数据支持。
免疫原性结果显示,加强后 28 天,mRNA 组针对原型株的中和抗体水平是灭活疫苗同源加强的 27 倍,针对奥密克戎 BA.1 变异株的中和抗体水平是灭活疫苗同源加强的 23 倍。
此外,交叉中和抗体动力学研究显示,序贯加强 1 剂康希诺生物新型冠状病毒 mRNA 疫苗后 7 天,针对当下流行的奥密克戎 BA.5 变异株的抗体水平即可达到峰值,是灭活疫苗同源加强的 29 倍。其中,60 岁及以上的老年人亚组也可诱导较高中和抗体,针对奥密克戎 BA.5 变异株的中和抗体水平是灭活疫苗同源加强的 23 倍。

境外创新药进展
境外部分,本周共有 17 款创新药(含改良新)研发进度推进到了新阶段,其中 1 款获批上市,3 款申报上市,5 款获批临床。
获批上市
卫材/渤健:
阿尔茨海默病新药 Aβ 单抗 Lecanemab 获 FDA 批准
1 月 7 日,FDA 加速批准由卫材(Eisai)和渤健(Biogen)联合开发的 Leqembi(lecanemab)用以治疗阿尔茨海默病(AD)!Leqembi 是近年来靶向 β 淀粉样蛋白的第二款创新阿尔茨海默病疗法,本次获批也代表在阿尔茨海默病治疗领域的又一项重要进展!
阿尔茨海默病是老年人中最常见的神经退行性疾病,而淀粉样蛋白沉积是患者大脑的标志性特征。靶向淀粉样蛋白是阿尔茨海默病新药开发的重要方向之一。Leqembi 为一抗 β 淀粉样蛋白(Aβ)抗体,能与可溶性 Aβ 聚合体结合,并且促进它们的清除。它具有改变疾病病理,缓解疾病进展的潜力,FDA 曾授予此疗法快速通道资格、优先审评资格、突破性疗法认定。
卫材和渤健在 2021 年下半年开始向 FDA 滚动提交上市申请(BLA),BLA 的提交基于 IIb 期概念验证临床试验 Study 201 的数据和 III 期临床试验 Clarify AD 的安全性数据。2022 年 5 月,他们完成了资料的滚动提交,以优先审评通道获 FDA 受理,给定的 PDUFA 决定日期在 2023 年 1 月 6 日。
2022 年 9 月 28 日,卫材/渤健共同宣布了确证性 III 期临床试验 Clarify AD 研究(试验登记号:NCT03887455)的大获成功。这一结果一经公布,就引起了轰动。除 Aducanumab 的 EMERGE 研究之外,这是第一个在确证性 III 期临床中取得积极结果的同类药物,具有里程碑意义。目前,这项结果的详细数据已经在 11 月的阿尔茨海默病临床试验(CTAD)会议上发布。
III 期临床试验 Clarity AD 结果 @Insight 数据库
 来自:Insight 数据库网页版
来自:Insight 数据库网页版
申报上市
Orasis Pharmaceuticals:
老花眼眼药水疗法 CSF-1 递交上市申请
1 月 4 日,Orasis Pharmaceuticals 公司宣布,已经向 FDA 递交其在研老花眼眼药水疗法 CSF-1 的新药申请(NDA)。CSF-1 是一款不含防腐剂的低剂量毛果芸香碱眼科溶液配方,有望为全球近 20 亿老花眼患者提供老花镜以外的治疗手段。
老花眼影响全球近 20 亿人,大多数患者只能选择佩戴老花镜,但是对患者来说并不方便。2021 年 10 月,艾伯维(AbbVie)旗下艾尔建(Allergan)公司的 Vuity(毛果芸香碱 1.25% 眼科溶液)获得美国 FDA 的批准,成为首款获批用于治疗老花眼的眼药水疗法。
Orasis 公司的 CSF-1 是一款由低剂量毛果芸香碱和多种载体构成的不含防腐剂的眼科溶液配方,其毛果芸香碱浓度为 0.4%。它的主要作用机制是收缩瞳孔,从而增强景深,改善近视力和中距视力。
这一 NDA 是基于包含超过 600 名患者的两项 3 期临床试验 NEAR-1 和 NEAR-2 的结果。两项 3 期临床试验在第 8 天均达到其主要终点和关键性次要终点,显著提高了视力表多读三行,并且远视力降低少于一行的患者比例。接受第一剂治疗后一小时内 40% 的患者多读三行,这一比例在接受第二剂治疗后达到 50%(p<0.0001)。受试者最早在接受治疗 20 分钟后就可获得视力改善,并且疗效可持续长达 8 个小时。
安全性方面,最常见的不良事件为头痛和滴注部位疼痛,分别出现在 6.8% 和 5.8% 的参与者中。在所有接受治疗的参与者中,只有 2.6% 报告出现中度治疗相关不良事件,其它为轻度不良事件。
阿斯利康/赛诺菲:
一针保护 5 个月!Beyfortus 上市申请获 FDA 受理
1 月 6 日,阿斯利康与赛诺菲联合宣布,FDA 接受其所开发 Beyfortus(nirsevimab)的生物制品许可申请(BLA),用以避免新生儿在他们第一个呼吸道合胞病毒(RSV)流行季节时,因感染所造成的下呼吸道疾病,以及避免 2 岁以下孩童在他们第二个 RSV 季节时的严重性 RSV 感染。FDA 预计在 2023 年第 3 季度公布审查结果。
RSV 是造成包含细支气管炎与肺炎等下呼吸道感染的最常见原因。它也是全世界导致婴幼儿住院的首要原因。在 2019 年,全球约有 3300 万急性下呼吸道感染病例,造成超过 300 万人住院,以及 26300 位 5 岁以下婴幼儿住院时死亡。
Beyfortus 是为所有婴幼儿设计的长效抗体疗法,从婴幼儿出生到第一个 RSV 季节只需一剂就能预防与 RSV 感染相关的疾病。作为一种单克隆抗体药物,Beyfortus 无需激活免疫系统便能提供及时、快速和直接的疾病免疫保护。此款在研药物曾获得 NMPA 与 FDA 的突破性疗法认定,以及欧洲药品管理局(EMA)的 PRIME 资格等。Beyfortus 在去年 11 月获得欧盟全球首次批准上市,用以避免新生儿在他们第一个呼吸道合胞病毒(RSV)流行季节时,因感染所造成的下呼吸道疾病。
Beyfortus 的上市申请是基于 MELODY 临床 3 期、MEDLEY 临床 2/3 期与一项临床 2b 试验的结果。
根据去年 3 月所公布的数据,MELODY 临床 3 期试验达主要终点,与安慰剂相比,Beyfortus 单次给药将 RSV 引起的需要接受治疗的婴幼儿下呼吸道感染(LRTI)发生率降低了 74.5%(95% CI:49.6-87.1,p<0.001)。
此外,MELODY 临床 3 期和 2b 期试验的预定汇总分析显示,推荐剂量的 Beyfortus 可减少 RSV 引起的住院。Beyfortus 的推荐剂量为:对于体重小于 5 公斤的婴幼儿为 50 mg 的单剂肌肉注射,而对于大于 5 公斤的婴儿,则为单剂肌肉注射 100 mg 的药物。
分析发现,在足月和早产婴幼儿中,安慰剂组 21/786 例婴幼儿(2.7%)和 Beyfortus 组 9/1564 例(0.6%)发生 RSV 相关住院,估计至给药后 150 天的疗效为 77.3%(95% CI:50.3-89.7,p<0.001)。MELODY 试验结果于 2022 年 3 月发表在著名医学期刊《新英格兰医学杂志》上。
临床试验结果披露
武田:
酶替代疗法 3 期临床试验结果积极
1 月 6 日,武田宣布,其酶替代疗法 TAK-755 在治疗先天性血栓性血小板减少性紫癜(cTTP)患者的关键性 3 期临床试验中获得积极结果,与目前标准治疗相比,将血小板减少事件发作频率降低 60%。基于这些数据,武田将向全球的监管机构递交上市申请。新闻稿指出,它有望成为首款治疗 cTTP 的 ADAMTS13 酶替代疗法。
cTTP 是一种超级罕见的慢性凝血疾病,发病原因是由于 ADAMTS13 酶的缺失,导致血管性血友病因子(VWF)多聚体在血液中积累。这些 VWF 多聚体的积累导致血小板的聚集和粘连,造成小血管中异常凝血,并出现溶血性贫血和血小板减少症。
新闻稿指出,TAK-755 是首款处于临床开发阶段的重组 ADAMTS13 蛋白,它通过替代缺失或功能失常的 ADAMTS13 酶,为 TTP 患者提供了一种靶向疗法。它已经获得美国 FDA 的快速通道资格,用于预防和治疗 cTTP。
这项临床试验评估了 TAK-755 与基于血浆的疗法(目前的标准治疗)相比,治疗 cTPP 患者的疗效和安全性。中期数据显示,TAK-755 与标准治疗相比,将患者出现血小板降低事件的频率降低 60%(95% CI,30%-70%)。此外,接受 TAK-775 治疗的患者中 8.9% 出现治疗相关不良事件,显著低于标准治疗(47.7%)。
多说一点
1 月 5 日,药明生物宣布与 GSK 达成许可协议,GSK 将获得基于药明生物专利技术平台开发的至多四款 TCE 双特异性/多特异性抗体的独家权利。
根据协议,GSK 将获得一款处于临床前阶段的双特异性抗体和其他至多三款处于早期发现阶段的 TCE 抗体的全球独家研究、开发、生产和商业化权利。该款双抗分别靶向肿瘤细胞表面的肿瘤相关抗原(TAA)和 T 细胞表面抗原 CD3,通过与肿瘤细胞及 T 细胞结合,特异性激活 T 细胞,从而杀伤肿瘤细胞,产生抗肿瘤活性。
药明生物将获得 4000 万美元首付款和最高达 14.6 亿美元的四款 TCE 抗体的研究、开发、注册和商业化里程碑付款。药明生物也有资格获得基于净销售额的分级销售提成。
免责声明:本文仅作消息分享,并不构成投资建议,也不代表 Insight 数据库的立场,文章观点仅供分享行业见解,请广大投资者谨慎。
编辑:Hebe
PR 稿对接:微信 insightxb
投稿:微信 insightxb;邮箱 insight@dxy.cn

点击阅读原文
免费试用 Insight 数据库






